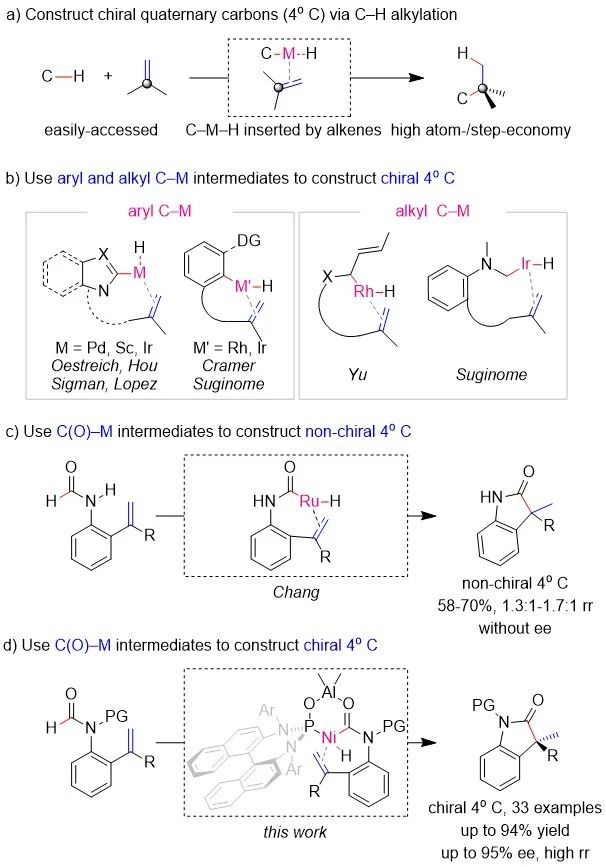
手性季碳中心普遍存在于众多天然产物、药物和农药中,其合成一直是有机合成的重点之一。虽然在现有的方法中,反应原料相对容易获得,反应过程中原子损失较小等使得对映选择性过渡金属催化的C-H烷基化成为构建手性季碳最具吸引力的合成方法之一(Scheme 1a),但是这一领域往往面临惰性C-H键反应活性较低,位阻较大的1,1-二取代烯烃难以插入C-M-H中间体以及区域选择性和对映体选择性难以控制等挑战。为解决这些问题,芳烃C-H键常常被用来构建季碳手性中心(Scheme 1b,左),因为这些C-H键活化后形成的C-M-H中间体能够被芳环稳定;另外,余和Suginome也实现了烯丙基或杂原子邻位的C-H键烷基化(Scheme 1b,右),而这些C-H键活化后形成的C-M-H中间体则能够被烯烃或杂原子稳定。通过以上分析不难发现,目前报道的合成策略主要是通过底物取代基来稳定C-M-H中间体,从而促进惰性C-H键活化和大位阻1,1-二取代烯烃的插入,最终构建了一系列手性季碳。虽然利用稳定的C-M-H中间体能够实现过渡金属催化C-H键烷基化构建手性季碳,但能否通过不稳定的C-M-H中间体来构建手性季碳仍然是一个未知的挑战。为此,Chang等人曾尝试通过Ru催化甲酰胺C(O)-H键烷基化来构建季碳。尽管他们成功实现了以季碳产物为主的区域选择性控制,但是最高仅1.7:1的选择性和消旋的产物一方面限制了反应的应用,另一方面表明甲酰胺C(O)-H键烷基化构建手性季碳是一个难以克服的挑战。不稳定的C(O)-M-H中间体会抑制生成手性季碳的原因主要是C(O)-M-H中间体脱羰释放的CO可能会阻碍手性配体与金属配位,从而使反应的区域选择性和对映选择性难以控制。为此,南开大学叶萌春课题组通过PO桥联的Ni-Al双金属催化实现了甲酰胺C(O)-H键与烯烃的烷基化反应,最终以单一的区域选择性和高达94%的收率及95%ee构建了一系列含手性季碳中心的吲哚酮类化合物。
作者以S1为模板底物对反应进行了初步探索,最终发现在没有外加Al的条件下,环化反应不发生,而外加20% mol AlMe3后,环化反应顺利进行并能以30%的收率和单一的选择性构建exo型环化的季碳产物。基于此结果,作者首先筛选了各种骨架衍生的PO配体,结果表明手性二胺和手性联萘酚衍生的PO对该反应没有活性(PO1-PO4),而虽然以TADDOL骨架衍生的PO为配体能够极大地提高反应活性(PO5),但是没有对映选择性控制。直到以手性联萘胺衍生的PO7作为手性配体时,反应能够以较低的ee值得到目标产物。据此,以连萘胺为基本手性骨架,作者探究了氮上的取代基(PO8-PO11)对反应的影响,最终发现3,5-二甲基苯基取代的PO10是反应的最优配体。在此最优配体的条件下,作者对其它常规条件进行了考察并发现,一方面Lewis酸对反应的影响较大,酸性稍弱的AlMe2Cl没有活性;另一方面,温度对反应也有较大的影响,室温为最佳温度,在此基础上升温或降温均会导致反应ee值下降;同时,0 ℃时的高反应活性表明目前的最优催化条件具有较高的反应性。

在最优条件的基础上,作者对底物的适用范围进行了考察。首先,作者考察了甲酰胺氮原子上保护基对反应的影响。实验结果表明反应对各种直链或支链的烷基取代基有良好的兼容性(1-10),以66%-94%的收率和78%-95%的ee值生成了目标产物。其中,含氧官能团的存在会导致反应的收率和ee值(6和9)略微降低,这一现象可能是O原子与Al配位引起的。另外,相较于烷基取代基,芳基取代基的引入会导致反应的收率和ee值(10-11)有所下降,原因可能是芳基取代基会降低羰基的电子云密度,从而抑制羰基与Al配位。同时,产物10的晶体结构表明手性季碳的绝对构型为R。除了甲酰胺氮原子上保护基,作者也考察了甲酰胺上的苯环取代基,结果表明虽然在氮原子的邻位和间位引入取代基均会导致反应的ee值下降(12-14),但是在对位引入供电子基(15-17)或者吸电子基(18),反应均能很好的发生,最终以87%-91%的收率和90%-92%的ee值合成了一系列手性季碳。随后,作者还研究了烯烃上的取代基对反应的影响,结果表明:反应对含有供电子基的芳基取代基具有较好的兼容性,如甲基(19和20)、烷氧基(21和22)和杂芳环(23-25),最终以76%-88%的收率和78%-93%的ee值构建了一系列手性季碳。值得注意的是,杂芳基环取代基的引入常常导致反应需要更多的AlMe3和更高的温度(23-25),这可能是因为杂原子的配位降低了AlMe3的Lewis酸性。与给电子基类似,反应对Cl(26)、F(27)和CF3(28)等吸电子基同样具有较好的兼容性,反应收率为61%-88%,ee值为85%-90%。另外,与芳基取代基相比,烷基取代基(29和30)仍然具有较好的反应效果,尽管反应ee值会有所下降(收率为64%-80%,ee值为52%-70%);可能是因为更柔性的烷基取代基会削弱催化剂对反应对映选择性的控制。值得一提的是,位阻很大的三取代烯(31),甚至四取代烯烃(32)在高温的反应条件下仍然能够以71%-77%的收率和53%-63%的ee值发生环化反应,这一结果表明催化体系具有较高的反应活性和兼容性。

为了证明当前反应的实用性,作者进行了相应的产物转化并合成了生物活性分子。首先,作者进行了模板反应的克量级实验并顺利地得到了目标产物1;虽然反应收率略有降低,但反应的ee值仍然保持(方案4a)。其次,产物1一方面可以被劳森试剂以71%的收率转化为硫代酰胺(33),另一方面可以被氢化铝锂以84%的收率还原为三级胺(34)。再者,通过BPO氧化和NaOH水溶液水解,产物中氮上的甲基保护基可以被很容易地脱除(35);随后的开环反应构建了含有手性季碳的氨基酸(36)。另外,产物1氮上取代基苯环的对位很容易被NBS溴化,生成相应的溴化产物37,从而能够实现以溴原子为连接点的进一步后修饰。最后,当前催化方法能够以81%的收率和90% ee合成一系列CCR9趋化因子受体拮抗剂的关键前体化合物38(方案4b)。

为了探究反应机理,作者进行了相关的机理实验。氘标记实验表明,甲酰胺的H完全转移到烯烃末端(Scheme 5a)。在平行实验中没有观察到明显的动力学同位素效应(kH/kD=1.04)(Scheme 5b),这表明甲酰胺C-H的活化没有参与决速步。此外,在反应体系中外加4-辛炔,同时将反应温度提升至80 ℃,反应除了以34%的收率和85%的ee值生成原环化产物1外,还以54%的收率和54%的ee值生成了新化合物39(Scheme 5c)。产物1和39的ee值不同,这一现象表明它们可能经历了不同的中间体。基于以上实验结果和以往研究,作者在Scheme 5d中提出了一个可能的反应机理:首先,S1与PO-Ni-Al双金属催化剂配位,得到了中间体A;随后中间体A会与烯烃直接进行分子内配体-配体氢转移(LLHT)从而形成中间体B,最后还原消除得到产物1。然而,若4-辛炔被加到反应体系中,生成的中间体A则可能直接与炔烃进行分子间配体-配体氢转移,进而形成中间体C,随后烯烃插入到C(O)-Ni键或Ni-C键中得到中间体D或D’,最后还原消除得到化合物39。

综上所述,作者在温和反应条件下,通过Ni-Al双金属催化的甲酰胺C(O)-H键与烯烃烷基化,以单一的区域选择性和高的对映选择性构建了含甲酰基的手性季碳。其中,反应成功的关键是联萘胺手性PO配体桥联的Ni-Al双金属催化剂;在此基础上,反应能够很好地兼容一系列二取代、三取代甚至四取代的烯烃,最终以高达94%的收率和95%的ee值合成了多种含有手性季碳的羟吲哚。另外,产物中酰胺基、吲哚酮和芳环取代基的转化实验表明了该方法的实用性。最后,机理实验揭示了Ni-Al双金属催化条件下,反应更有可能通过LLHT机理来构建季碳手性中心。
相关研究成果发表于Angewandte Chemie International Edition。南开大学化学学院元素有机化学国家重点实验室的博士研究生王豪锐为该文第一作者,南开大学化学学院元素有机化学国家重点实验室叶萌春教授和博士研究生徐魏魏为该工作的通讯作者。相关工作得到国家重点研发计划、国家自然科学基金委等项目的支持。
原文(扫描或长按二维码,识别后直达原文页面):

Enantioselective Construction of Oxindoles Bearing a Quaternary Carbon via Ni–Al Bimetal-Catalyzed Formyl C–H Alkylation
Haorui Wang, Jiang-Fei Li, Mengying Xu, Qi-Lin Zhou, Weiwei Xu, Mengchun Ye
Angew. Chem. Int. Ed., 2024, DOI: 10.1002/anie.202413652

