自19世纪Fischer开创不对称合成领域以来,不对称催化得到了迅速发展并广泛用于合成手性药物、农药以及精细化学品。具体而言,不对称催化是通过手性催化剂与底物的前手性中心或前手性面之间的精确识别来控制立体选择性。然而,当前手性中心或前手性面上两个基团的空间位阻和电性相似时(如二烷基酮类化合物),手性催化剂则难以区分。相比之下,生物催化中的次级相互作用(如氢键、静电/偶极相互作用、蛋白质芳香残基的π-π堆积)可确保酶和底物之间的准确识别,从而产生优异的立体选择性。受生物催化模型(特别是锁-钥模型)的启发,化学家开发出各种策略来控制立体选择性(Ber. Dtsch. Chem. Ges., 1894, 27, 2985–2993; J. Am. Chem. Soc., 2013, 135, 12480–12496),例如:为特定的不对称反应量身定制的手性催化剂(图1a)。
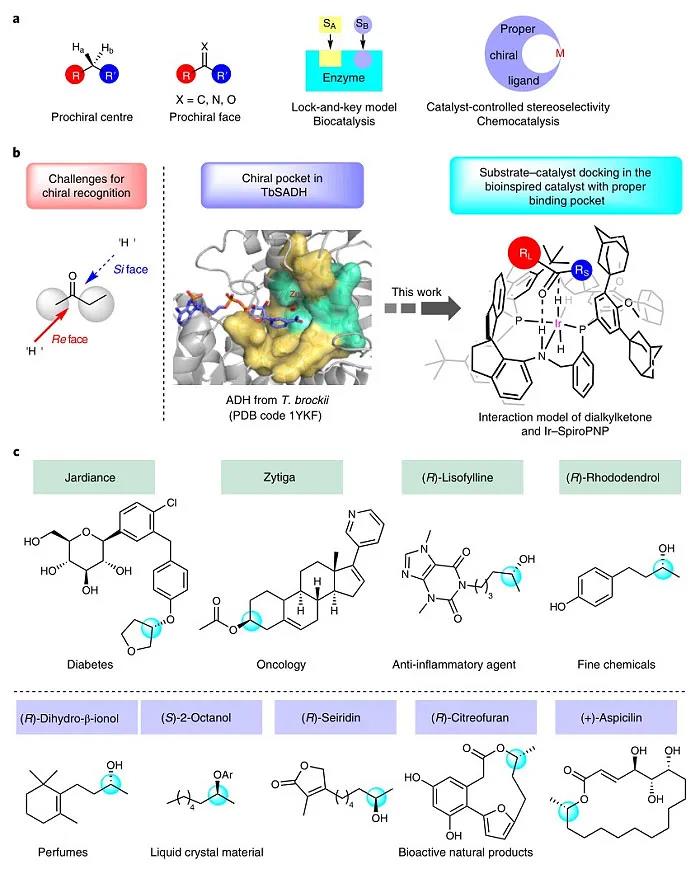
图1. 二烷基酮的催化不对称氢化。图片来源:Nat. Catal.
1986年,Keinan等人通过X-射线衍射证实了来自布氏嗜热厌氧菌的醇脱氢酶在结构上有一个小口袋和一个大口袋(J. Am. Chem. Soc., 1986, 108, 162–169),该酶活性位点的大小和形状赋予其在酮的还原反应中具有较高的底物特异性和优异的立体选择性。受此启发,南开大学周其林院士团队设想能否通过具有拥挤且狭窄的手性口袋的催化剂来精确区分二烷基酮的两个非常相似的烷基(图1b)。近日,他们设计并合成了具有三齿手性螺膦-胺-膦配体(SpiroPNP)的铱催化剂,成功地实现了二烷基酮的对映选择性氢化反应。相关成果发表在Nature Catalysis 上。
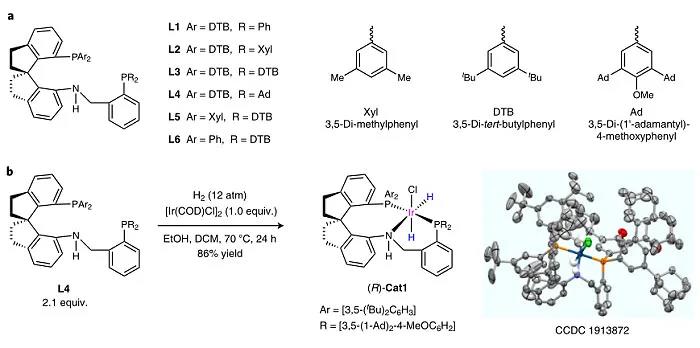
图2. spiroPNP 配体和Ir–spiroPNP催化剂的制备。图片来源:Nat. Catal.
首先,作者从相应的螺氨基膦合成了六个SpiroPNP配体(命名为L1-L6;图2a)。为了评估这些配体的性能,作者用原位生成的铱催化剂来进行2-癸酮(S7)的氢化反应。结果显示L4(Ar = 3,5-二叔丁基苯基,R = 3,5-二-(1'-金刚烷基)-4-甲氧基苯基)的对映选择性最高(78% e.e.),这表明增加配体的空间位阻能提高反应的对映选择性。随后,作者在氢气气氛下将[Ir(COD)Cl]
2
与L4混合,以86%的收率分离出铱催化剂(R)-Cat1(图2b)。
1
H NMR、
31
P NMR和高分辨质谱表明(R)-Cat1具有两个负氢并以cis-形式连接到铱原子上。单晶X-射线衍射分析表明活性中心非常拥挤。如图1b所示,根据金属-配体双功能机理,催化剂中的NH基团对于酮的氢化反应至关重要。值得一提的是,催化剂(R)-Cat1非常稳定,即使在空气中储存一个月,其纯度和反应活性没有任何变化,这归因于手性配体的三齿螺环结构和庞大的膦基将其活性中心包围。
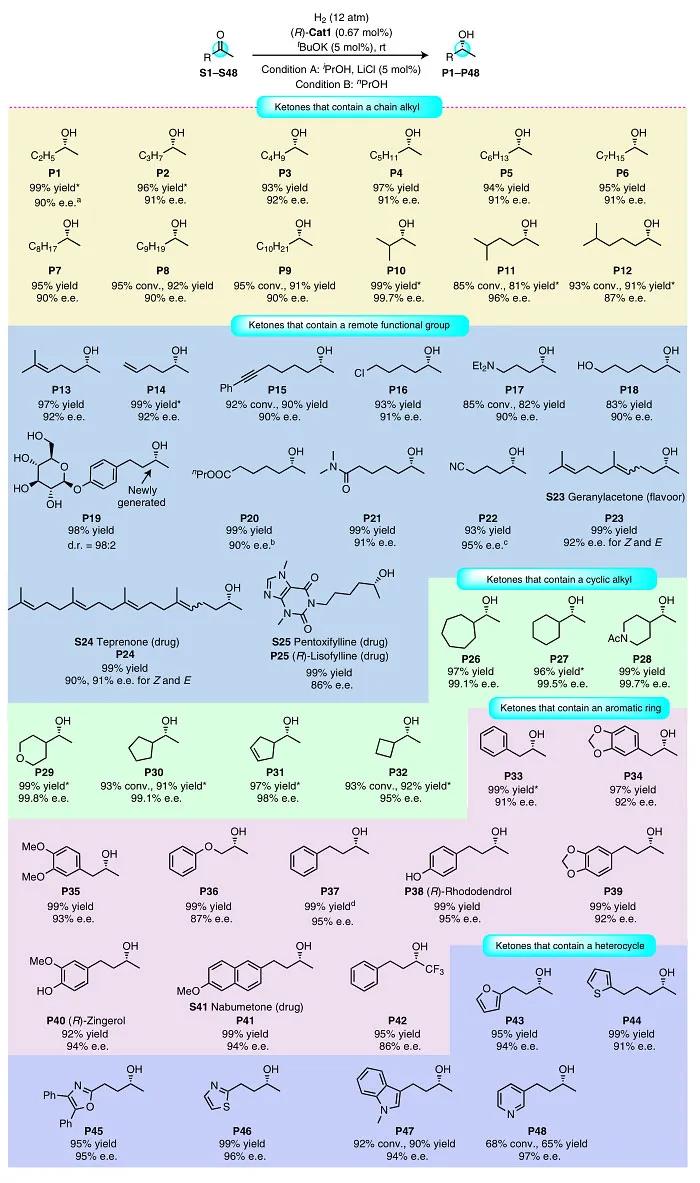
图3. 底物扩展一。图片来源:Nat. Catal.
当作者使用(R)-Cat1催化S7的氢化反应时,对映选择性提高到88% e.e.,对反应条件(H
2
压力、溶剂和添加剂等)进行优化,e.e. 值有所提高(90%)。在最佳条件下,作者考察了二烷基酮的底物范围(图3)。首先是烷基甲基酮类底物(最具挑战性的酮底物之一),结果显示在室温下便可将带有甲基和更长链烷基的酮(S1-S12)氢化,以85-100%的转化率和87-99.7%的e.e. 值得到相应的醇,并且烷基链的长度对立体化学的影响不大。值得一提的是,最具挑战性的底物2-丁酮(S1)在室温下进行氢化反应时,以88%的e.e. 值得到2-丁醇(P1),而在0 °C下进行反应时,e.e. 值为90%,这表明(R)-Cat1可以区分底物的甲基和乙基。然而,在相同的反应条件下,(R)-Cat1不能还原芳香酮(如苯乙酮)。
接下来,作者考察了远程带有官能团的酮。带有烯烃、炔烃、卤原子、氨基、羟基、糖基、酯基、酰胺和氰基的底物都能兼容该反应,以优异的转化率(85–100%)和对映选择性(90–95%e.e.)得到目标产物(S13–S22)。由于修饰生物活性分子是新药研发的有效策略,因此作者对天然香料化合物香叶基丙酮(S23)和胃溃疡药物替普瑞酮(S24)进行了氢化反应,并以优异的对映选择性和收率得到氢化产物。值得一提的是,该方法可以一步法将己酮可可碱(S25)氢化获得商用抗炎药(R)-赖索茶碱(P25),而先前的报道则需要冗长的步骤(J. Am. Chem. Soc., 2014, 136, 17426–17429)。此外,环烷基甲基酮(S26-S32)也能进行氢化反应,以极高的对映选择性(95-99.8%e.e.)得到仲醇——药物合成(如组蛋白甲基化酶EZH2抑制剂)的重要合成子。同样地,芳基丙烷-2-酮(S33-S36)和芳基丁烷-2-酮(S37-S42)也能高对映选择性地进行氢化反应,得到手性1-芳基-2-丙醇和4-芳基-2-丁醇,它们是许多手性药物的基元,例如:醇P34是合成抗癫痫药物talampanel的关键中间体,醇P36是合成Dibenzyline的原料。值得一提的是,在S37的氢化反应中,催化剂(R)-Cat1的负载量可以低至0.02 mol%,并且循环使用六次也不会降低催化活性和对映选择性。此外,带有杂芳环的酮(呋喃、噻吩、噁唑、噻唑、吲哚和吡啶)都能实现氢化反应,以优异的对映选择性得到手性醇(P43-P48)——合成各种天然产物和药物的起始原料。

图4. 底物扩展二。图片来源:Nat. Catal.
鉴于手性环醇(如3-羟基哌啶)是许多药物的基元,也是天然产物的重要合成子,因此作者考察了各种脂肪族环酮(图4),均能以高收率和高对映选择性获得相应的环醇。如在0.1 mol% (R)-Cat1的存在下,以克级规模制备商用抗癌药物(R)-ibrutinib(2018年零售额为62.05亿美元)的活性成分——(S)-N-Boc-3-羟基哌啶(P52,1.4 g,收率:99%,e.e. 值:90%),而先前的报道则需要多步过程(Helv. Chim. Acta, 2014, 97, 1507–1515; Org. Process Res. Dev., 2014, 18, 827–830)。另外,通过动态动力学拆分对2-甲基环己酮进行不对称氢化,以98%的e.e. 值(trans/cis = 5:1)得到trans-2-甲基环己醇(P55)作为主要产物。当用于类固醇(S56-S59)的氢化反应时,也能以极好的收率(99%)和非对映选择性(> 99:1 d.r.)得到目标产物。
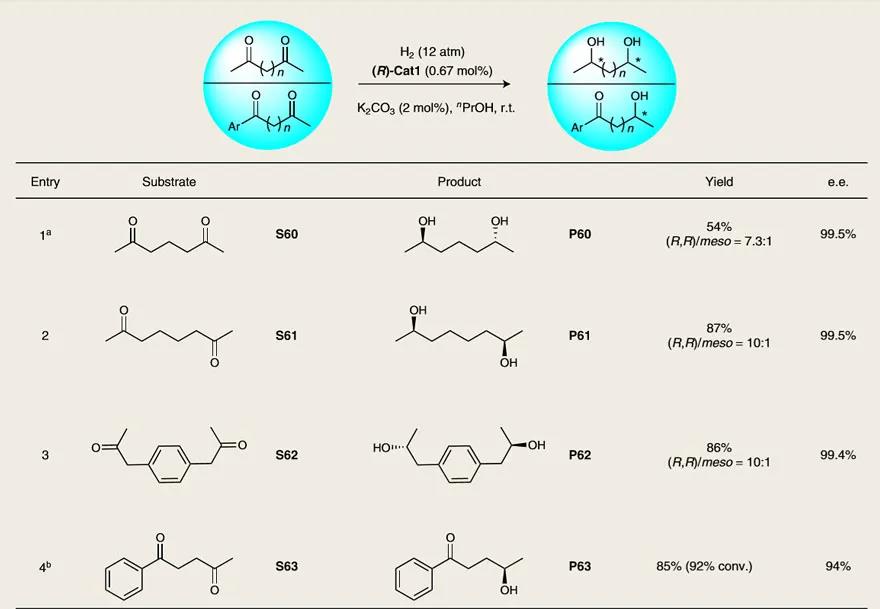
图5. 二酮的不对称氢化。图片来源:Nat. Catal.
由于手性二醇是合成各种药物和手性双膦配体的重要起始原料,因此作者探究了二酮类化合物的不对称氢化反应(图5)。2,6-庚二酮(S60)能以良好的收率(54%)、优异的对映选择性(99.5% e.e.)和良好的非对映选择性((R,R)/meso = 7.3:1)得到手性1,5-二醇P60——用于制备2,6-二取代的六元杂环(存在于许多药物中,如吡美诺、氯吡酰胺和抗菌剂)。类似地,S61和S62也能兼容氢化反应,以较好的收率和非对映选择性得到手性二醇。值得注意的是,(R)-Cat1能选择性地催化二酮S63中脂肪族酮的氢化反应,得到具有抗癌活性的酮醇P63。
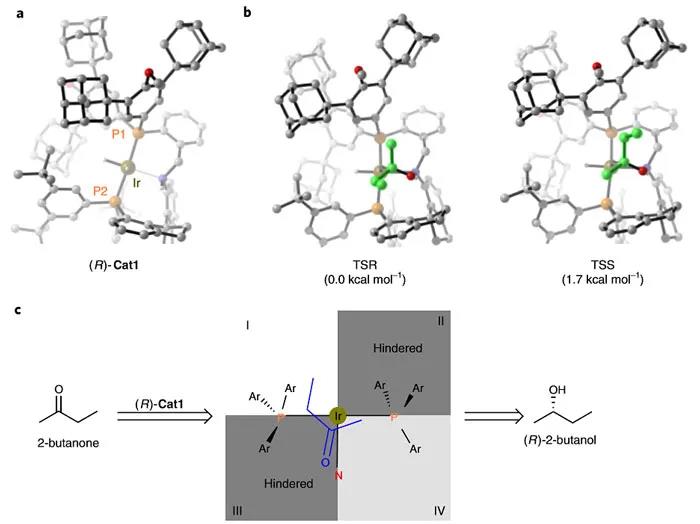
图6. DFT计算。图片来源:Nat. Catal.
为了探究反应对映选择性的起源,作者对(R)-Cat1催化的2-丁酮氢化反应进行了密度泛函理论(DFT)计算。(R)-Cat1的球棍模型表明P2原子上的3,5-二叔丁基苯基、P1原子上的3,5-二金刚烷基以及配体的螺环骨架构成了一个深而狭窄的手性口袋(图6a)。由于该口袋具有拥挤的手性环境,当底物靠近催化剂的活性中心时,其方向会受到限制。计算表明R产物过渡态(TSR)的能量要比S产物过渡态(TSS)的能量低1.7 kcal mol
-1
(图6b),这与该反应得到R产物的实验结果一致。此外,作者还利用象限模型来解释2-丁酮不对称氢化反应中(R)-Cat1的对映体控制(图6c)。象限II和III分别被具有金刚烷基和螺双茚满的芳基所占据。象限IV中的芳基与金刚烷基连接,使其比象限I中的芳基(与叔丁基相连)更拥挤,这允许底物以si-面接近催化剂,从而形成了R产物。
南开大学周其林院士团队报道了二烷基酮的对映选择性氢化反应,该反应的关键在于三齿手性螺膦-胺-膦配体(SpiroPNP)的铱催化剂,它具有狭窄、拥挤的手性口袋。该方案不仅解决了不对称催化氢化反应中的长期难题,也为手性药物和精细化学品的合成提供了一条简单、高效的方法。
原文:
Enantioselective hydrogenation of dialkyl ketones
Feng-Hua Zhang , Fa-Jian Zhang, Mao-Lin Li, Jian-Hua Xie , Qi-Lin Zhou
Nat. Catal
., 2020, DOI: 10.1038/s41929-020-0474-5

