近日,南开大学陈弓教授、何刚特聘研究员团队以3,3'- di-F-BINOL为手性配体和2-甲氧基-5-氯碘苯为氧化剂通过PdII-催化/喹啉导向的3-芳基丙胺的分子内不对称C(sp3)-H酰胺化实现了β-芳基-β-内酰胺的不对称合成,该成果发表于近期ACS Catal.(DOI: 10.1021/acscatal.9b04768)。
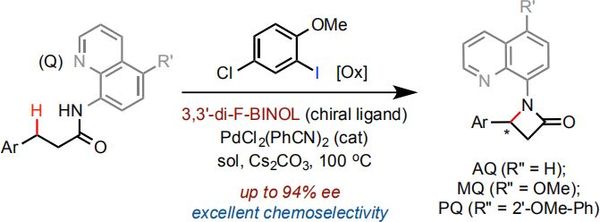
钯催化的定向C(sp3)-H官能团化已经成为构建各种脂肪族骨架的重要手段,酰胺键连接的二齿辅基导向基团在新键生成中显示出其独特的优势即高反应性和广泛应用性。然而,由于金属中心缺乏合适的配位点,这些二齿辅基的络合方式给对映体控制造成困难,但最近的研究表明这种对映体诱导是可能的(Scheme 1A)。段伟良课题组报道了基于BINOL的手性磷酸或酰胺配体/PdII-催化下氨基喹啉(AQ)导向的3-芳基丙酰胺与芳基碘化物之间的苄基β-C-H芳基化,该反应表现出中等至良好的对映选择性;随后,史炳锋课题组开发了通过3,3'-di-F-BINOL/PdII-催化下吡啶基异丙基胺(PIP)导向的3-烷基丙酰胺与炔基溴之间的对映选择性β-C(sp3)-H烷基化反应。为了进一步扩展二齿辅基介导的C(sp3)-H官能团化的实用性,需要开发新的对映选择性反应模式。
最近,吴斌课题组报道了PdII-催化下AQ导向的3-烷基和3-芳基丙酰胺之间的β-C-H官能团化反应,当加入过量的五氟碘苯作为氧化剂时可以高收率和化学选择性得到β-内酰胺产物(Scheme 1B)。作者认为,-C6F5强吸电子基团可以抑制PdIV中间体的C-C还原消除(RE),从而促进分子内C-N RE。近日,南开大学陈弓教授、何刚特聘研究员团队以3,3'- di-F-BINOL为手性配体和2-甲氧基-5-氯碘苯为氧化剂通过PdII-催化/喹啉导向的3-芳基丙胺的分子内不对称C(sp3)-H酰胺化实现了β-芳基-β-内酰胺的不对称合成。
β-内酰胺不仅是药物设计中的重要骨架,也是有机合成中的重要中间体。长期以来,主要通过金属催化的分子内C-H卡宾插入而进行的C-H官能团化策略构建β-内酰胺。近年来,Pd-催化的分子内C(sp3)-H官能团化包括C-H烷基化、羰基化胺化、通过Pd0/II或PdII/0催化循环的甲氨酰化等为构建β-内酰胺提供了新途径。在这些反应中,基于C-H烷基化和甲氨酰化的对映选择性转化也分别用手性膦酸盐和磷酰胺配体进行了验证。相比之下,以烷基酰胺底物的C-H酰胺化为特征的催化反应可以提供更直接的切断策略。这种类型的外消旋转化是通过在氧化条件下促进高价金属中间体的C-N RE来实现的。为了使该转化具有对映选择性,需要开发合适的手性配体和氧化剂组合。
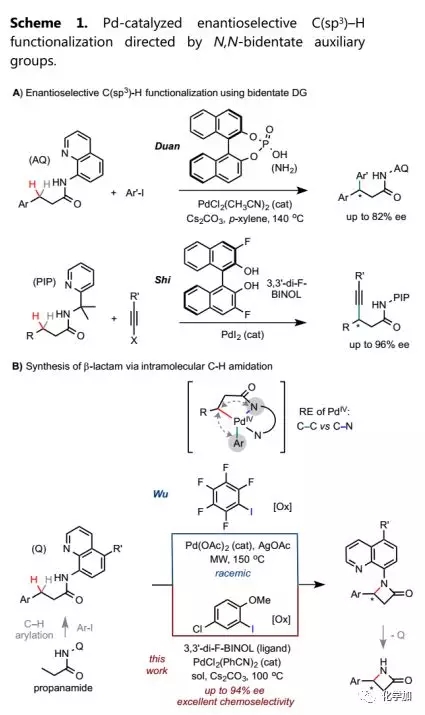
最近,作者对PdII-催化下AQ导向的3-苯基丙酰胺1与芳基碘化物之间的对映选择性C(sp3)-H芳基化再研究中注意到,利用手性磷酸酯配体可以不同的收率和对映选择性得到β-内酰胺2作为副产物(Table 1)。尽管五氟碘苯I-1氧化剂在外消旋体系(Pd(OAc)2作为催化剂、AgOAc作为I-清除剂、150 ℃反应)中具有最佳的反应性和化学选择性,但在100 ℃的PdCl2(CH3CN)2、Cs2CO3和手性磷酸酯配体条件下反应性较差。通过对缺电子ArI氧化剂的考察发现,3,5-di-CF3-苯基碘化物(I-2)表现出的化学和对映选择性最佳。利用I-2(4 eq.)、20 mol% 3,3'-二芳基(3,5-di-CF3-C6H3)取代的BINOL衍生的磷酸酯配体L4和 30 mol% dba的100 ℃无溶剂条件(conditions A)可以59%的收率得到2(85% ee)并伴有15%的C-H芳基化产物3。此外,喹啉辅基的结构对反应性和ee值也有影响,利用AQ的C5位甲氧基类似物Q-2(MQ)可以显著提高内酰胺产物的收率和ee值。除缺电子的芳基碘化物外,作者发现含各种邻位取代基的富电子芳基碘化物也可以不同程度地促进2的形成。例如,含邻位甲基的I-10只得到环化产物,但反应性低;含两个邻位甲氧基的I-8可以58%的收率和中等ee值得到2。

当用手性BINOL配体时,对于富电子和缺电子的ArI氧化剂均产生中等到良好的对映选择性(Table 2)。与间位含甲氧基的I-4和I-12(PhI)相比,邻位含甲氧基的I-5得到2的收率更高。在I-5的C5位引入吸电子基团得到的I-6和I-7进一步改善了化学选择性,作者选择I-6进一步优化,其中di-F-BINOL配体L-9和L-6组合效果最佳。在10 mol% PdCl2(PhCN)2/20 mol% L-9催化(conditions B)下,1与I-6(4 eq.)在100 ℃ 1,3-di-CF3-Ph/tAmOH混合溶剂中反应以90%的收率得到2(89% ee),并伴有6%的芳基化副产物。在B'条件下,使用MQ与AQ得到的结果相当(1 vs 7)。利用在AQ的C5位引入邻甲氧基苯基的Q-9(PQ)得到的内酰胺产物的ee值最高(94%)。还值得注意的是:1)Cs2CO3对于获得高ee值至关重要;2)加入银添加剂如Ag2CO3可以使ee值显著降低;3)Pd(OAc)2催化剂产生的ee值较低;4)要获得高收率需要4 eq. ArI。

在最佳反应条件B下,作者考察了在AQ、MQ和PQ辅基下的底物范围(Scheme 2):通过Pd-催化下连接相应辅基的丙酰胺前体与芳基碘之间的单选择性β-C-H芳基化制备原料3-芳基丙酰胺。辅基的性能因β-芳基的不同而略有波动。通常,芳基的间位和对位被取代后,仍具有良好的耐受性;邻位被取代或缺电子芳基的收率和ee值显著降低。如13所示,在标准条件下,非活化C(sp3)-H键的分子内胺化具有非常低的收率和中等的ee值。
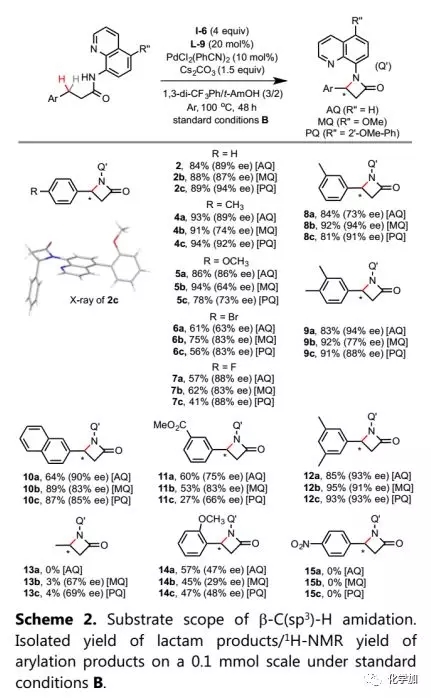
连接MQ的丙烯酰胺16与Phl通过Pd-催化的单选择性β-C-H芳基化及后续β-C-H胺化得到化合物2b(Scheme 3A),然后通过CAN脱除MQ得到β-内酰胺产物17(收率75%,87% ee)。如19所示,通过铜介导的区域选择性碘化和钯催化的Suzuki偶联很容易在AQ的C5位引入Ar''基团。通过Pd-催化下PQ导向的β-C-H芳基化和β-C-H胺化得到化合物2c,其PQ也可以通过CAN脱除从而以中等收率得到17(94% ee),经过己烷/乙酸乙酯重结晶后,其ee值提高至99%。通过Boc对17酰胺化并经LiOH裂解可以两步75%的收率得到苯基取代的β-氨基酸20(99% ee);将17与芳基碘通过Hartwig-Buchwald偶联可以90%的收率得到21。

(图片来源:ACS Catal.)
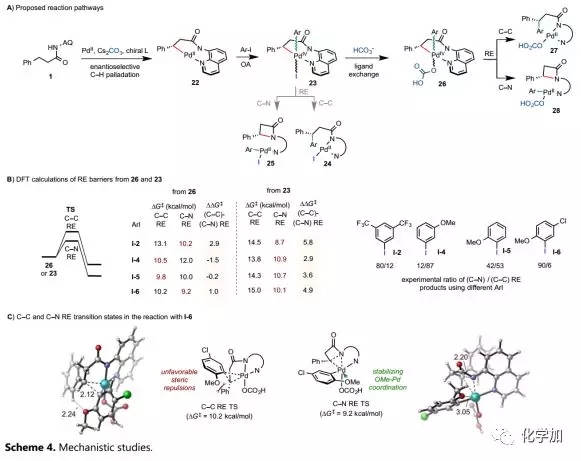
这种Pd-催化的氨基喹啉导向的分子内C-H酰胺化是按照C-H钯化、氧化加成和还原消除的顺序进行(Scheme 4A)。与其他PdII-催化的C(sp3)-H官能团化一样,基于BINOL的磷酸酯或di-F-BINOL配体控制的C-H钯化(形成PdII-钯环22)可能是该反应的对映体决定步骤;与ArI氧化加成后,PdIV中间体23经C-C或C-N RE分别得到芳基化24或环化产物25;经过原位脱钯化和I-配体的解离可再生活性PdII-催化剂。为了了解ArI氧化剂对化学选择性的作用,作者与匹兹堡大学的刘鹏教授合作,通过密度泛函理论(DFT)计算研究模型底物1与各种芳基碘化物反应中的C-C和C-N RE过渡态(TS)。前期对Pd-催化的ArI的C-H芳基化的计算研究表明,I-键合的PdIV中间体可能在RE前经过阴离子配体交换,作者认为23可能与原位生成的HCO3-反应形成新的PdIV中间体26。因此,通过计算考虑了23和26的还原消除,并且计算的26的C-C vs C-N RE的化学选择性与实验结果一致(Scheme 4B)。相比之下,计算的I-键合23的RE的化学选择性,特别是I-4的化学选择性显著偏离了观察到的比率。与吴斌课题组报道的外消旋体系中使用的C6F5-I(I-1)氧化剂类似,含两个强吸电子CF3基团的I-2通过电子效应不利于PdIV的C-C RE从而促进内酰胺的形成。含邻位甲氧基的富电子芳基碘化物(I-5和I-6)的内酰胺产物的选择性提高可能是由于ArI的邻位甲氧基和β-Ph之间的空间位阻使CC RE过渡状态不稳定所致(Scheme 4C)。在C-N RE过渡态中,弱的o-OMe-Pd配位可能进一步促进C-N键的形成。在I-5的C5'位引入Cl原子可以微调I-6的电性和对C-N RE的选择性。以上计算分析表明,利用邻位甲基取代的I-10对内酰胺具有良好的化学选择性和低反应性,前者是由于抑制C-C RE的类似空间效应所致,后者是由于最初的OA难以转化为钯环所致。
小结:南开大学陈弓、何刚团队首次报道了PdII-催化的分子内不对称C(sp3)-H酰胺化方法,可以由丙酸和芳基碘化物前体不对称合成β-芳基β-内酰胺,其ee值最高可达94%。其中,2-甲氧基-5-氯碘苯氧化剂对于实现其高化学选择性至关重要。机理研究表明,非常规芳基碘化物氧化剂的空间和电子效应共同控制了PdIV中间体的C-N和C-C还原消除途径的竞争。此外,内酰胺产物的喹啉辅基可以通过温和的条件脱除。

