近日,南开大学谢建华课题组与天津大学黄跟平课题组和浙江九洲药业严普查团队合作,发展了“添加酯基活化底物”的策略,并利用超高效手性螺环铱催化剂Ir-SpiroPAP首次实现了喹啉的部分不对称催化氢化,从而为手性1,4-二氢喹啉的不对称合成提供了绿色、高效合成新方法。相关成果已在线发表在Journal of the American Chemical Society 上,该论文的第一作者是南开大学大学博士研究生朱昌亮。该研究成果得到国家重点研发计划项目及国家自然科学基金的经费支持。
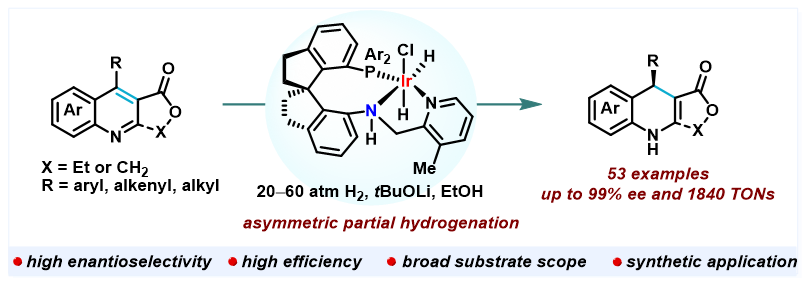
发展高效、高选择性的不对称催化新反应和新方法是有机合成化学研究领域的热点和挑战。手性1,4-二氢喹啉是一类重要的含氮杂环化合物,也是有机合成中重要的手性原料。传统的合成方法通常依赖于手性试剂的多步合成,而不对称催化合成方法虽然在近年来得到了快速发展,但主要仅限于有机小分子催化的不对称环加成反应和不对称转移氢化,以及铜催化烷基镁试剂的不对称加成。喹啉的不对称催化氢化是合成手性氢化喹啉最为绿色和原子经济性方法。自2003年周永贵等实现首例喹啉的不对称催化氢化合成手性四氢喹啉以来,喹啉的不对称催化氢化备受关注,但实现其部分不对称氢化仍是挑战。关键问题在于喹啉经催化氢化失去芳香性后生成了更为活泼的1,4-二氢喹啉中间体,容易继续被氢化形成手性四氢喹啉。因此,实现喹啉的部分不对称催化氢化的选择性控制非常困难,极具挑战性。
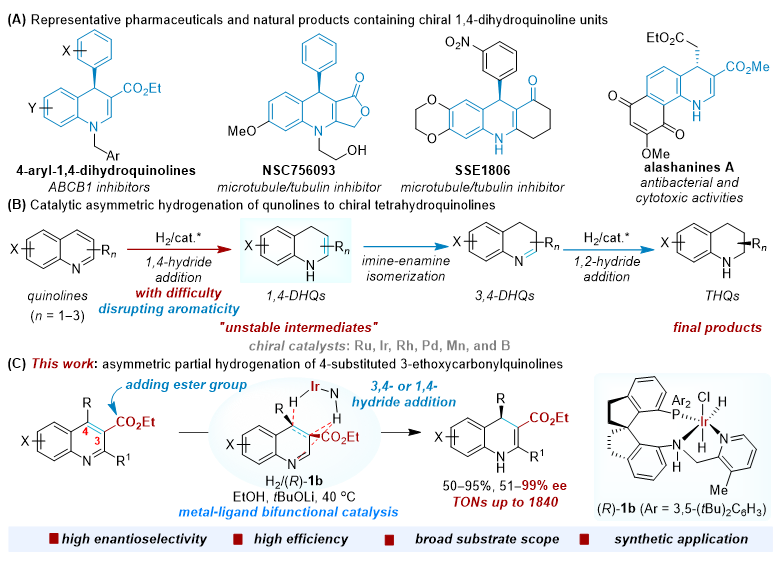
图1. 喹啉的不对称催化氢化和部分不对称催化氢化
作者在前期的研究中已发展了超高效手性螺环铱催化剂Ir-SpiroPAP,实现了简单酮、酮酸酯、消旋酯及缺电子烯烃等的高效、高选择性不对称催化氢化反应,并成功将这些高效不对称催化氢化新反应应用于手性药物及天然产物的不对称合成(Acc. Chem. Res. 2023, 56, 332−349)。在此基础上,作者为解决喹啉部分不对称催化氢化选择性控制困难的问题,设计了“添加酯基活化底物”的策略。具体而言,在喹啉环3-位添加酯基,从而增加C3-C4双键的缺电子性和极性,并利用手性螺环铱催化剂Ir-SpiroPAP通过金属-配体双官能化机理,实现负氢的3,4-或1,4-加成到喹啉环C4位,打破喹啉的芳香性。随后通过亚胺-烯胺异构化或直接得到手性1,4-二氢喹啉。值得注意的是,由于所得1,4-二氢喹啉中烯胺C-C双键的极性较低,手性螺环铱催化剂Ir-SpiroPAP对其表现一定的惰性,从而有效解决喹啉部分不对称氢化选择性控制困难的挑战。
作者通过对手性螺环催化剂Ir-SpiroPAP进行系统的评价和反应条件优化,发现在叔丁醇锂(40 mol%)作碱,乙醇为溶剂及50 atm和40 ℃反应的条件下,吡啶环3-位甲基取代的手性螺环铱催化剂(R)-1b(0.5 mol%)对模型底物4-苯基-3-乙氧羰基喹啉(2a)的部分不对称催化氢化,可以以93%的收率和95% ee的对映选择性得到(R)-4-苯基-3-乙氧羰基-1,4-二氢喹啉((R)-3a)。在优化反应条件下,作者对底物范围进行了拓展:1)对3-乙氧羰基喹啉底物,喹啉环4-位的取代基可以为芳基、杂芳基、芳烯基和烷基,对映选择性可高达99% ee,且烯基双键保持不变(图2);2)对呋喃酮并喹啉类底物,即9-取代呋喃[3,4-b]喹啉-1(3H)-酮4,反应活性略微降低,但当9-位取代基为芳基、杂芳基和烷基取代时,依然能够获得高收率和高对映选择性,最高可达99%的ee值(图3)。此外,喹啉环C2位有取代基,如甲基时,虽然反应活性显著降低,但也可以给出好的对映选择性。
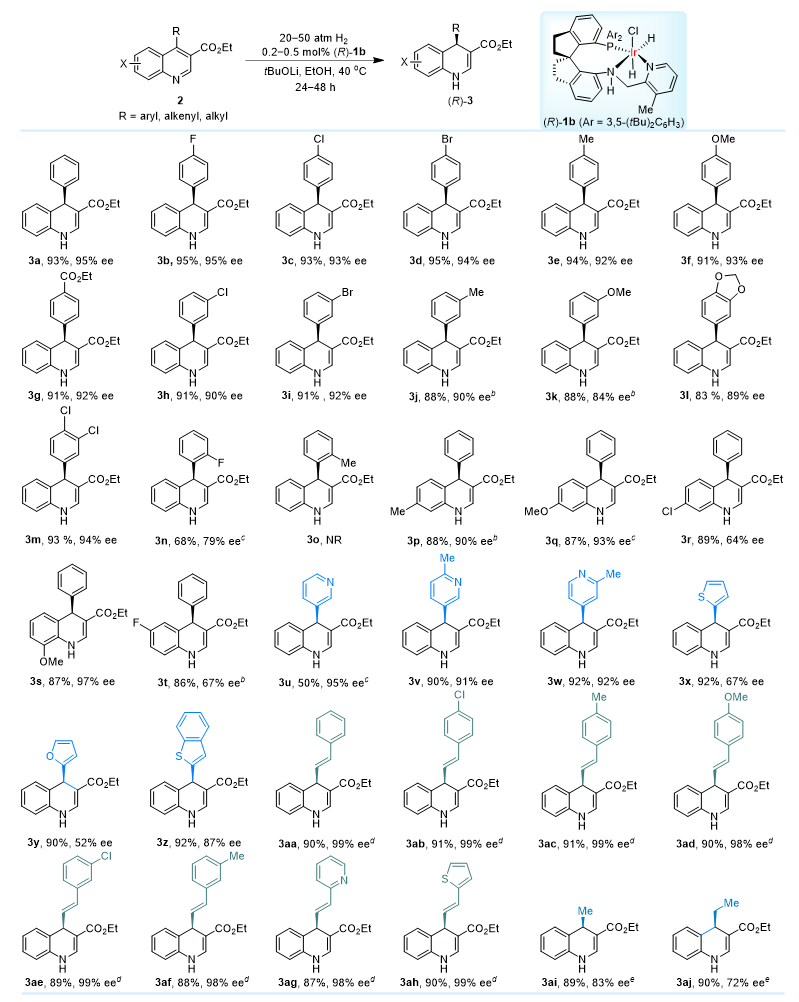
图2. 4-取代3-乙氧羰基喹啉的部分不对称催化氢化底物范围
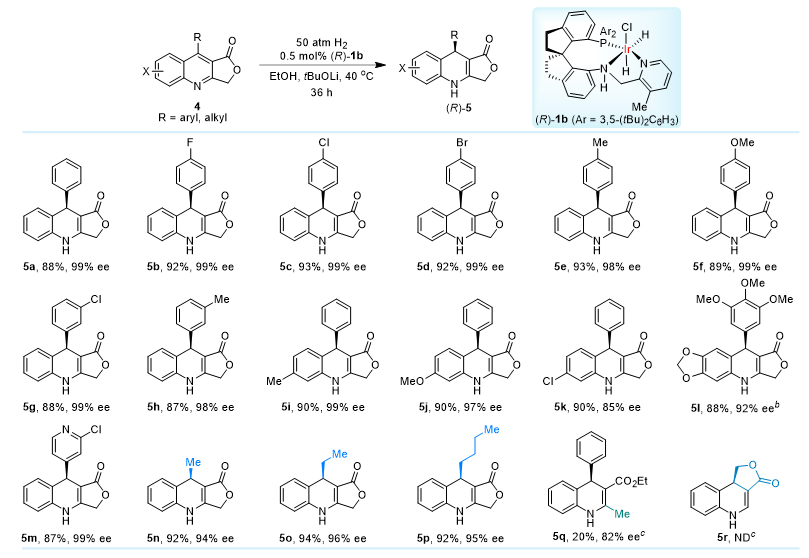
图3. 9-取代呋喃[3,4-b]喹啉-1(3H)-酮的部分不对称催化氢化底物范围
在研究氢化反应的应用转化过程中,作者将催化剂用量降低至0.05 mol%成功实现了所选择性底物的克级不对称催化氢化,获得了高收率和基本保持不变的对映选择性以及达到1840的高转化数。在此基础上,作者进一步对将氢化产物进行了转化,成功实现了杂氮杂鬼臼毒素类似物NSC750212和NSC756093,以及褪黑激素MT2受体激动剂等目标化合物的不对称合成(图4)。
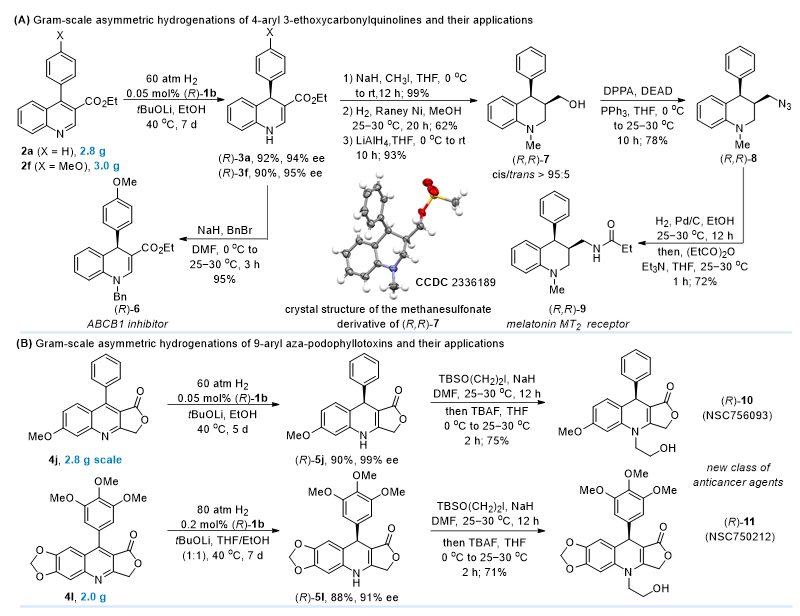
图4. 克级规模不对称氢化及应用转化
在机理探索实验研究中,作者发现3-位添加吸电子取代基是实现氢化反应的关键,没有取代基时不会发生氢化反应;氘代实验表明,铱催化剂中的负氢是加成到喹啉环C4位;跟踪实验表明,在d8-甲苯为溶剂时,观察到3,4-二氢喹啉12和烯醇盐13的存在,尝试分离鉴定它们以及将氢化产物(R)-3a转化为3,4-二氢喹啉12和烯醇盐13却为成功;竞争性Hammett实验展示,4-位苯环上对位为供电子取代基(如Me、MeO)对反应速度无影响,但为吸电子取代基(如Cl、F)则显著加快反应速度(图5)。通过DFT计算研究,揭示氢化反应不是通过经典的金属-配体促进的六元环过渡态协同机理进行的,而是通过分步机理经历了负氢转移和H2分子的断裂。而铱催化剂中的N-H与底物形成N-H---O氢键和N-H---π相互作用,稳定过渡态进而提高反应的立体选择性控制(图6)。
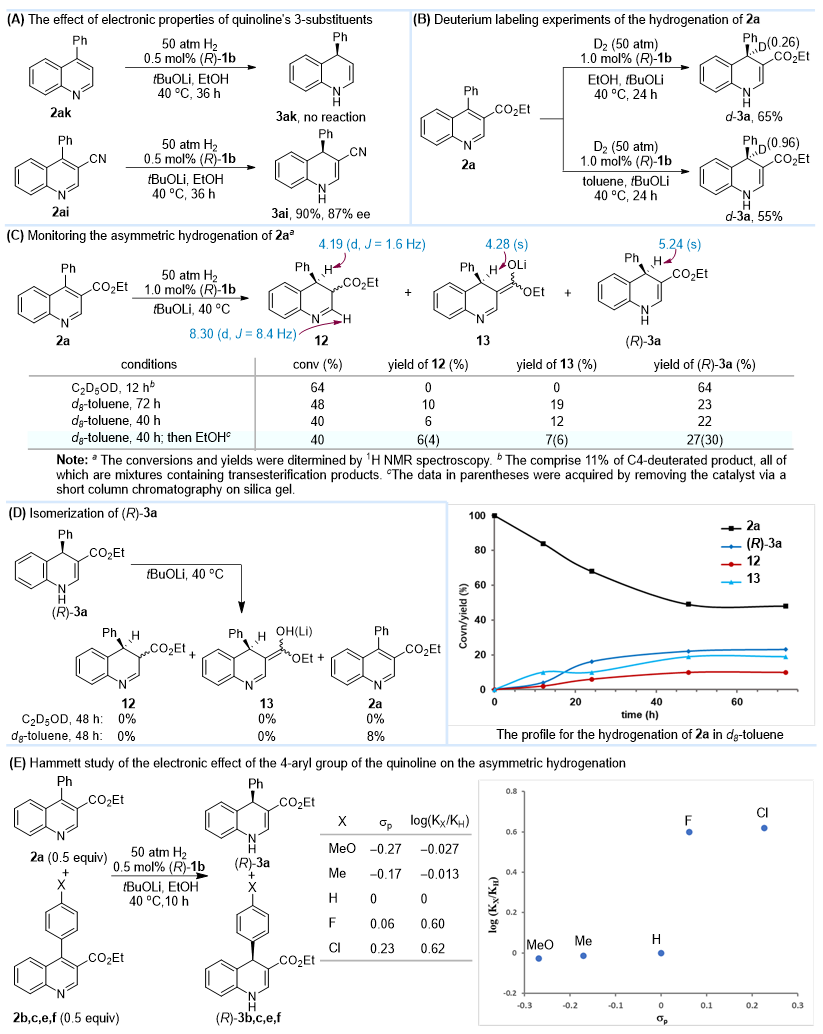
图5. 机理研究实验
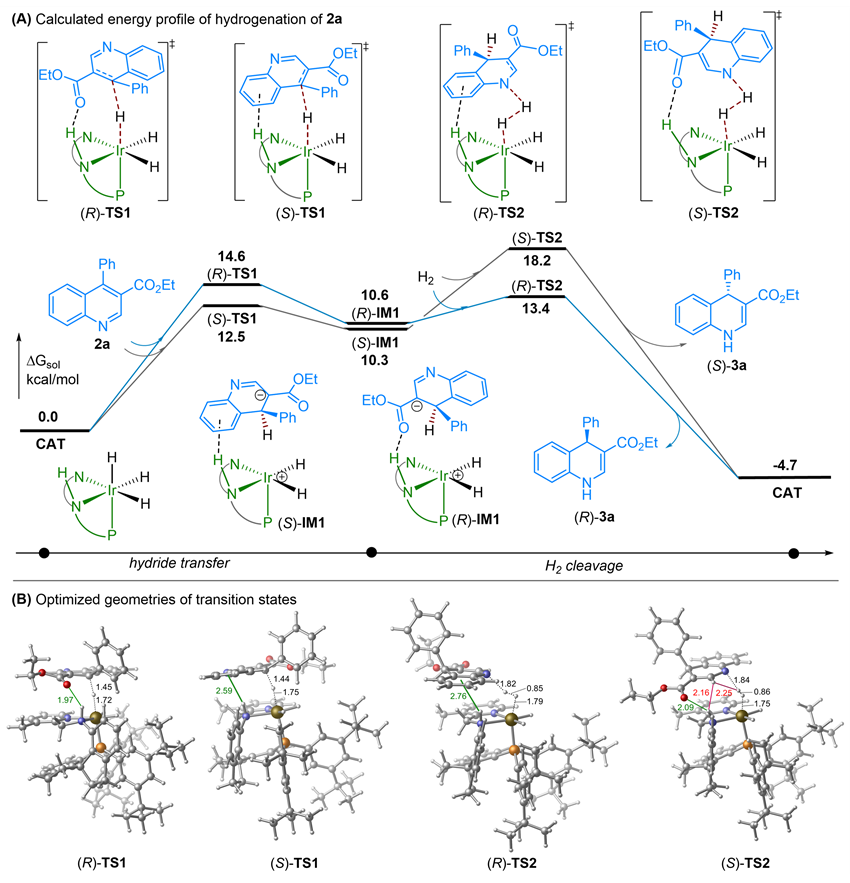
图6. DFT计算的可能机理
综上所述,作者通过“添加酯基活化底物”的策略,并利用手性螺环吡啶胺基膦配体的铱催化剂Ir-SpiroPAP,成功实现了首例喹啉的部分不对称催化氢化合成手性1,4-二氢喹啉。该催化剂表现出极高的活性和效率,对系列4-取代-3-乙氧羰基喹啉及其呋喃酮并喹啉类似物表现了高达99% ee的对映选择性和达到1840的高转化数,为手性1,4-二氢喹啉的不对称合成提供了绿色、高效新方法。机理研究揭示,喹啉环3-位添加酯基等吸电子基团是实现喹啉部分不对称催化氢化的关键。氢化过程不是通过经典的金属-配体双官能化的六元环过渡态协同机理进行,而是经历了负氢的转移然后经H2分子断裂的分步机理。
原文(扫描或长按二维码,识别后直达原文页面):

Enantioselective Synthesis of Chiral 1,4-Dihydroquinolines via Iridium-Catalyzed Asymmetric Partial Hydrogenation of Quinolines
Chang-Liang Zhu, Xueyuan Yan, Huai-Yu Bin, Xiong Wu, Zheng-Yan Huang, Pu-Cha Yan*, Genping Huang*, Jian-Hua Xie*, Qi-Lin Zhou
J. Am. Chem. Soc. 2025, DOI: 10.1021/jacs.4c13618

