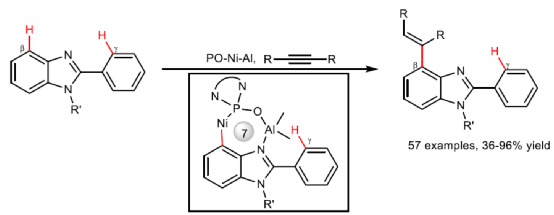
炔烃与非活化C(sp2)-H键通过螯合C-H键金属化进行的氢芳化反应,主要发生在γ-位(涉及形成稳定的五元金属环),而对于β-C(sp2)-H键的氢芳化反应则具有挑战。近日,南开大学叶萌春课题组开发了一种膦氧配位Ni-Al双金属催化体系,从而实现了炔烃与β-C-H键的氢芳化反应,涉及形成罕见的七元环镍中间体。文章链接DOI:10.1038/s41467-022-30367-8
(图片来源:Nat. Commun.)
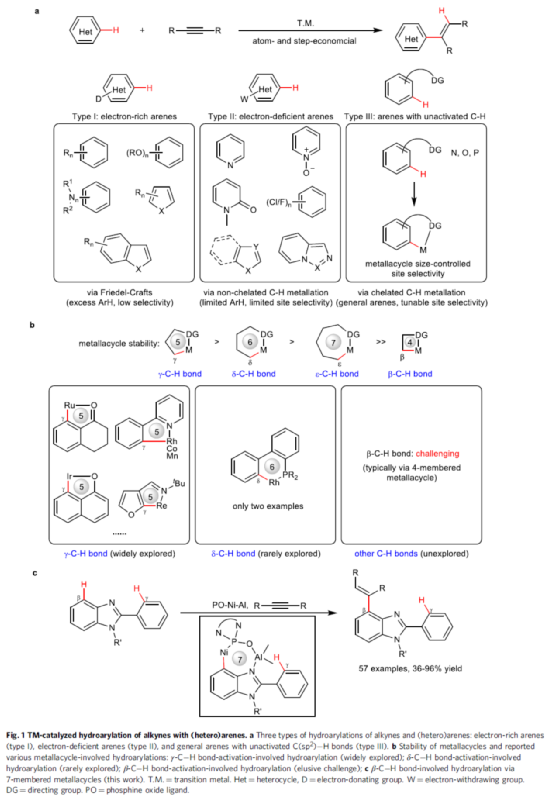
炔烃与芳烃的氢芳化反应是合成芳基烯烃(广泛存在于天然产物、生物活性化合物和材料分子中)的一种具有高原子经济性和步骤经济性的策略。在过去的几十年里,化学家已开发出多种过渡金属催化氢芳基化反应的策略。根据芳烃的电子性质,氢芳化可分为三种类型(Fig. 1a):富电子(杂)芳烃(Type I),缺电子(杂)芳烃(Type II)以及具有非活化C(sp2)-H键的一般芳烃(Type III)。其中,Type I反应主要通过Friedel-Crafts型途径进行,需多个富电子取代基来提高(杂)芳烃的电子密度,从而导致底物范围有限和位点选择性困难。相反,大多数type II反应是通过氧化加成途径进行,因为底物中存在强负电性的杂原子或吸电子基团导致电子密度分布不均,从而使缺电子的C-H键很容易与金属发生氧化加成。然而,独特的电性需求使得底物仅限于特殊的杂环化合物,如吡啶、多氟芳烃、咪唑等。为了实现非活化C(sp2)-H键与芳烃的氢芳化反应,已开发向底物中引入适当的导向基团用于螯合C-H键金属化的设计(Type III)。在导向基团的协助下,非活化C(sp2)-H键可被金属化,并且通过调节所形成金属环的尺寸原则上可实现不同位点的选择性。基于这些优势,螯合C-H键金属化参与的氢芳化反应在过去几十年中通过已得到了广泛的探索(Fig. 1b, left)。然而,大多数例子仅限于γ-位C-H键与导向基团中的配位原子反应,因为形成的稳定五元金属环比其他较大(六元或七元)或更小的(四元)金属环具有更有利的熵效应和环张力。目前,仅有两例涉及通过六元金属环参与δ-C-H键氢芳化反应的例子(Fig. 1b, middle)。相比之下,对于其他涉及C-H键氢芳化反应却很少被研究(Fig. 1b, right)。在此,南开大学叶萌春课题组开发了一种膦氧配位Ni-Al双金属催化体系,从而实现了炔烃与β-C-H键的氢芳化反应,涉及形成罕见的七元环镍中间体(Fig. 1c)。
(图片来源:Nat. Commun.)
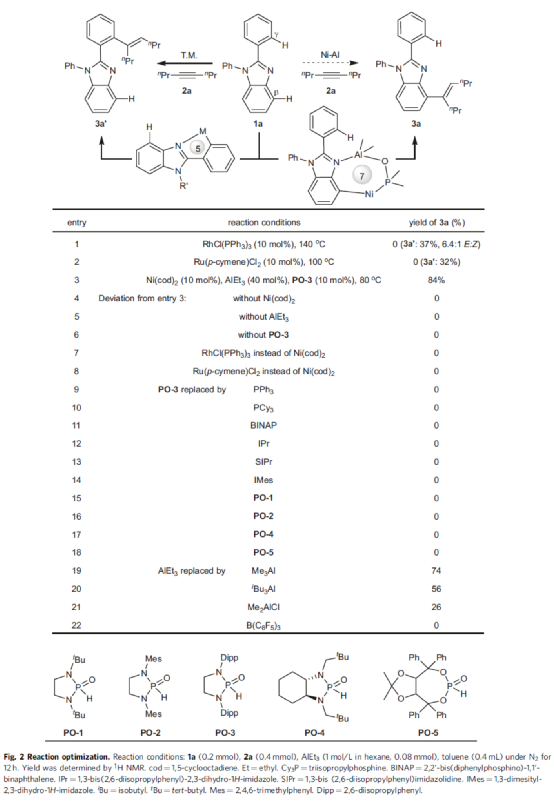
首先,作者以2-苯基苯并咪唑1a与4-辛炔2a作为模型底物,进行了相关氢芳化反应条件的筛选(Fig. 2)。当以Ni(cod)2作为催化剂,PO-3作为配体,AlEt3作为Lewis酸,在甲苯溶剂中80 oC下反应,能以84%的收率获得产物3a。
(图片来源:Nat. Commun.)
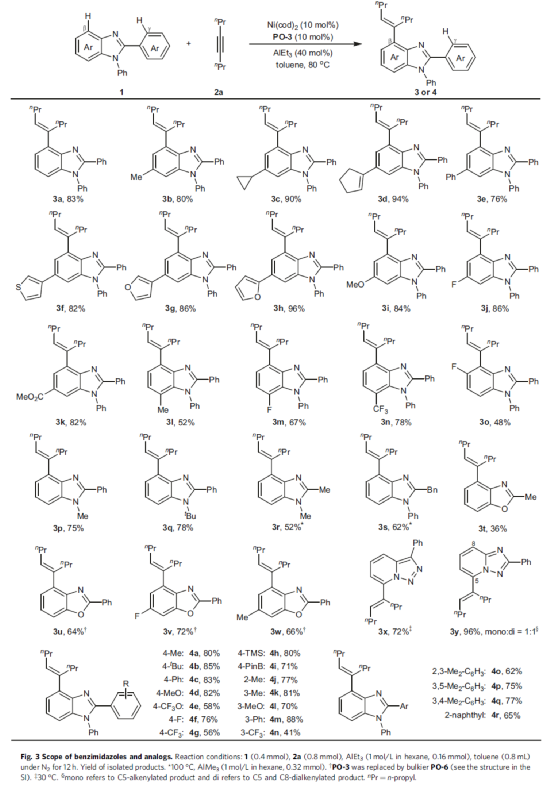
在获得上述最佳反应条件后,作者首先对2-苯基苯并咪唑的底物范围进行了扩展(Fig. 3)。首先,在底物中的C6-位含有烷基、烯基、芳基、杂芳基、甲氧基、酯基、卤素等时,均可顺利反应,获得相应的产物3b-3k,收率为76-96%。其次,在底物中的C7-位含有甲基时,可获得52%收率的产物3l。当底物中的C7-位含有缺电子氟基(3m)和三氟甲基(3n),可将收率分别提高至67%和78%。然而,当底物中的C5-位含有氟基(3o)时,由于空间位阻导致收率偏低(48%)。当底物中的N1-位含有甲基、叔丁基时,均可顺利反应,获得相应的产物3p-3q,收率为75-78%。当底物中的C2-位含有甲基、苄基时,需提高反应的温度以及增加Al-Lewis酸的负载量,可使反应顺利进行,获得产物3r-3s,收率为52-62%。除了苯并咪唑,其他的杂环化合物可在优化条件下顺利反应,如苯并噁唑(3t-3w)和三唑(3x和3y)。此外,当底物中的C2-位含有一系列不同电性取代的芳基与萘基时,也均为合适的底物,获得相应的产物4a-4r,收率为41-88%。

(图片来源:Nat. Commun.)
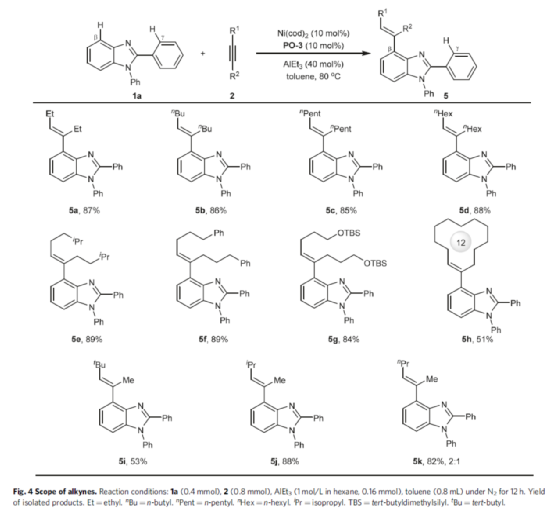
紧接着,作者对炔烃底物范围进行了扩展(Fig. 4)。首先,一系列不同取代的炔烃底物,如乙基、正丁基、正戊基、正己基、异己基等时,均可顺利进行反应,获得相应的产物5a-5g,收率为84-89%。同时,环状炔烃也为合适的底物,获得51%收率的产物5h。其次,对于一些非对称的炔烃底物,也与体系兼容,获得相应的产物5i-5k,收率53-88%,但区域选择性很大程度上取决于炔烃取代基的空间位阻。例如,叔丁基甲基炔烃(5i)和异丙基甲基炔烃(5j)可生成单一的区域异构体产物,而正丙基甲基炔烃(5k)则生成比例为2:1的区域异构体混合物。
(图片来源:Nat. Commun.)
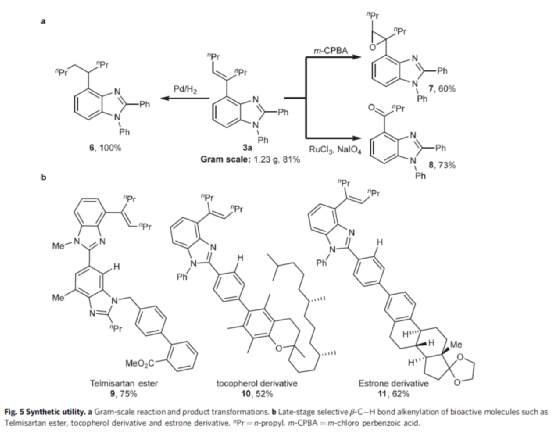
随后,作者对反应的实用性进行了研究(Fig. 5)。首先,克级规模实验,同样能够以81%收率得到产物3a。其次,3a中的烯基可通过氢化与氧化反应,获得相应的化合物6-8,收率为60-100%。此外,该策略还可用于生物活性分子的后期修饰,如替米沙坦酯(9)、生育酚(10)和雌酮(11)衍生物.
(图片来源:Nat. Commun.)
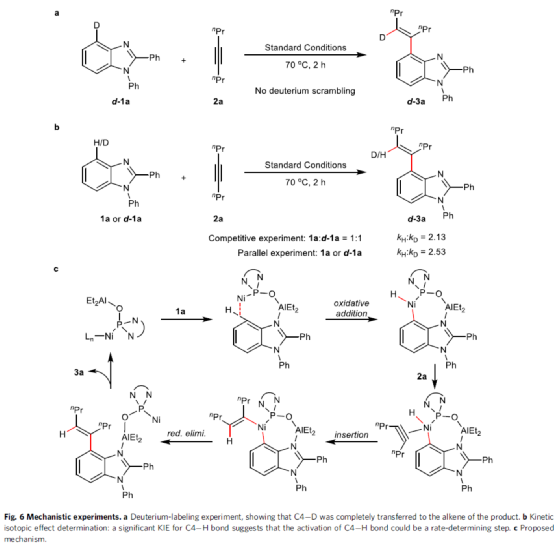
为了进一步了解反应的机理,作者进行了相关的实验研究。首先,通过d-1a与2a的氘标记实验发现,烯基上的氢完全来自苯并咪唑C4-位的芳基氢,从而表明C4-H键发生金属化(Fig. 6a)。其次,通过KIE实验表明,C4-H键的断裂可能是决速步骤(Fig. 6b)。基于上述的研究以及相关文献的查阅,作者提出了一种可能的催化循环过程(Fig. 6c)。首先,膦氧配位的Ni-Al双金属催化剂与咪唑中的N原子配位,从而形成七元环镍中间体。随后,经氧化加成、插入和还原消除的过程,从而获得目标产物3a并再生双金属催化剂。
(图片来源:Nat. Commun.)
总结:南开大学叶萌春课题组报道了一种膦氧配位Ni-Al双金属催化炔烃与β-C-H键的氢芳化反应,从而合成了一系列C4-烯基化的2-苯基苯并咪唑衍生物,具有底物范围广泛、收率高、化学选择性出色等特点。值得注意的是,膦氧配位Ni-Al双金属催化能够有效地引导镍通过七元金属环进行β-C-H键的氢芳化反应,从而避免了高张力四元镍环的形成。

