
2019年11月22日,南开大学周其林及朱守非团队在《Science 》在线发表题为“Highly enantioselective carbene insertion into N–H bonds of aliphatic amines”的研究论文,该研究报道了基于两种催化剂串联的高对映选择性卡宾插入脂肪胺N–H键合成手性氨基酸的策略。此项研究不仅解决了对映选择性卡宾插入反应的长期挑战,而且为涉及强配位底物的过渡金属催化的不对称转化提供了潜在的通用策略。哈佛大学Eric N. Jacobsen在同期《Science 》发表了题为"A catalytic one-two punch"的评论文章,对该项研究进行了系统解读,并给予了高度评价。
手性胺在天然产物,药物和农用化学品中几乎无处不在。2016年,Top200的处方药中约43%含有脂肪族胺部分。因此,过渡金属催化的高对映选择性C–N键形成反应一直以来都是合成化学家关注的内容。在这方面,过渡金属催化的卡宾插入到N-H键已被证明是一种简单的方法,具有温和的反应条件、良好的官能团耐受性和易于获得的反应物。近来,手性过渡金属催化剂已经成功地应用到对映选择性天然或非天然α手性氨基酸衍生物的合成(N-H插入反应)。然而,这些反应仅限制于芳族胺或酰胺(图1B )。因为脂族胺是一种相对更强的路易斯碱,会通过与金属催化剂配位造成中毒,从而干扰金属卡宾的形成,这为N-H插入反应构成了巨大挑战。此外,过量的脂肪胺可能会取代金属叶立德中间体中的叶立德,从而导致游离叶立德形成外消旋产物(图1C,上)。
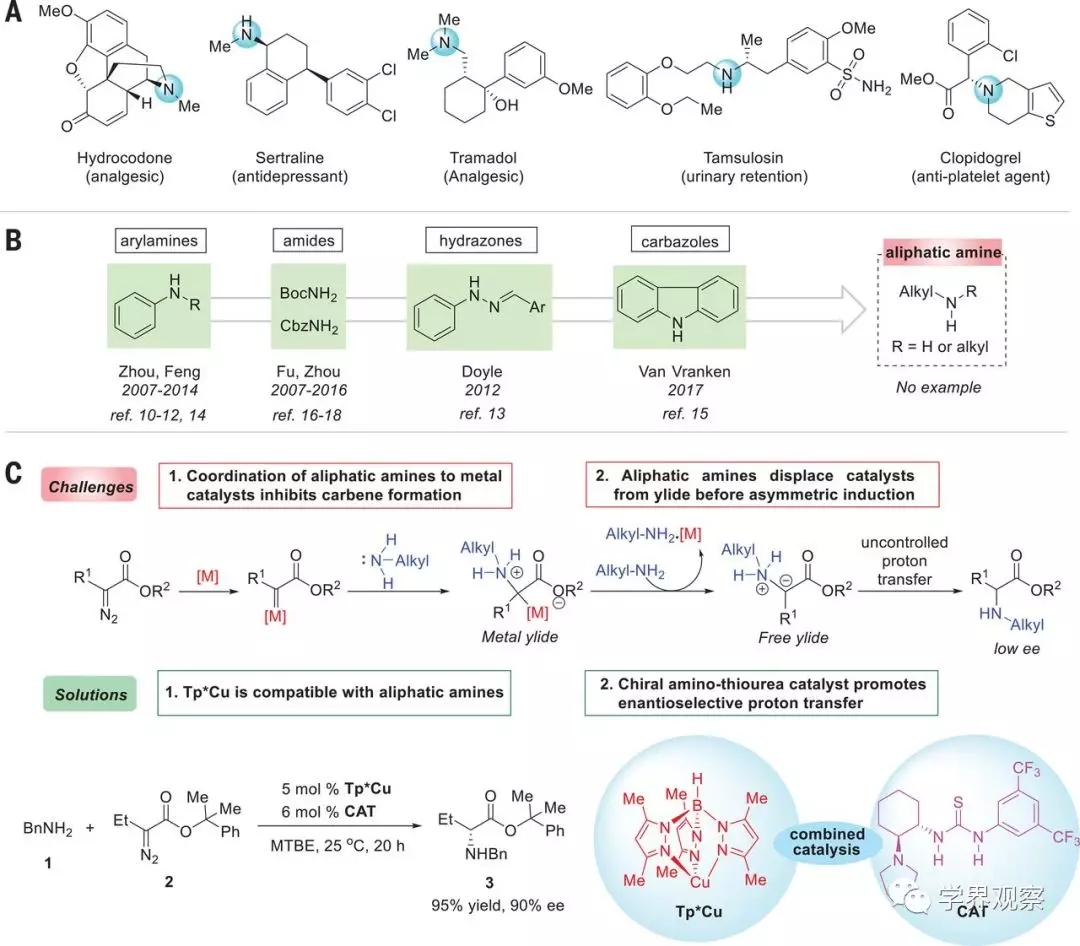
图1 对映体控制脂肪胺与卡宾的N–H插入反应策略。(A)含有手性脂肪胺的代表性药物。(B)已报道的对映选择性NH插入反应的胺源。(C)合成脂肪族胺的对映选择性过渡金属催化的N–H插入反应。最佳反应条件:1(0.2 mmol),2(0.22 mmol),Tp*Cu(5摩尔%)和CAT(6摩尔%)的反应在3 ml甲基叔丁基醚(MTBE)中在25°C下放置20小时。
南开大学周其林院士和朱守非教授等人设想两种催化剂的组合可能会解决这些挑战:与脂族胺相容的非手性过渡金属催化剂将生成叶立德中间体,然后单独的手性催化剂将促进对映选择性质子转移。通过探索α-重氮丁酸酯与苄胺N-H插入反应中的各种过渡金属催化剂和手性H键催化剂组合,作者最终找到了最佳的反应条件(图1C,下),为很难用其他方法制备的手性α-烷基α-氨基酸衍生物提供了高效、高对映选择性的合成方法。

图2 在对映选择性N–H插入反应中脂肪胺和α-重氮酯的范围。(A)脂族胺的范围。(B)在药物的对映选择性后期功能化中的应用。(C)α-重氮酯的范围。
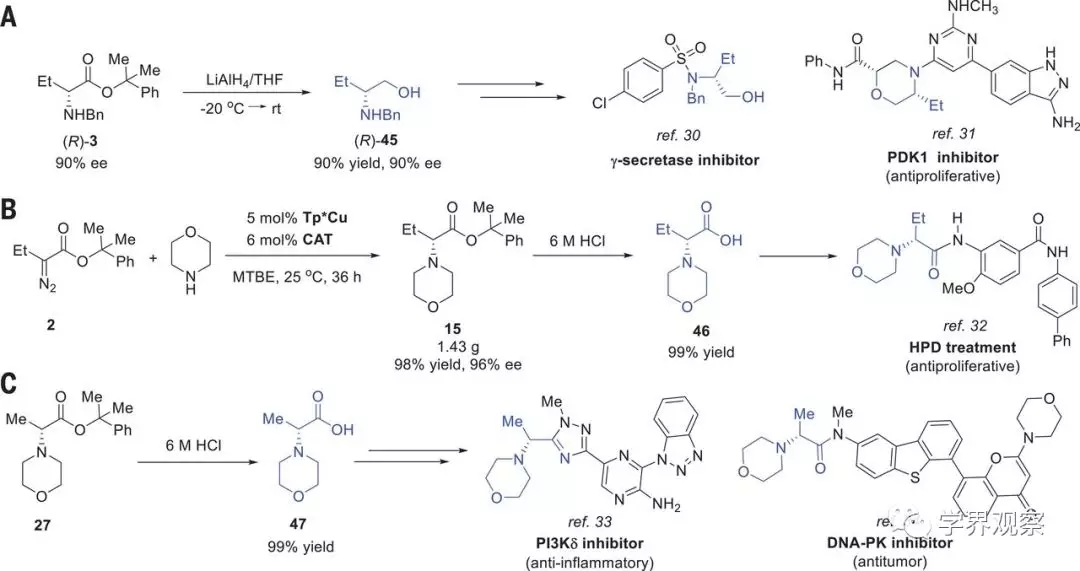
图3 NH插入产物的合成转换。(A)(R)-3通过LiAlH4向(R)-45 转化,合成具有抗肿瘤活性的生物活性分子(PDK1抑制剂)的关键中间体。(B)以NH插入为关键步骤合成HPD。(C)27向47转化,合成生物活性分子(PI3K5抑制剂)的关键中间体。
为了更深入地了解NH插入反应的机理,作者使用在线红外(IR)光谱进行了动力学分析。在40°C下以各种浓度的组分测量了反应的初始速率。该速率显示出Tp*Cu和重氮化合物2浓度的一级依赖性(图4A),这表明Tp*Cu催化的重氮酯2形成金属类化合物是可能的限速步骤。然而,通常与金属催化剂配合并抑制金属卡宾形成的苄胺在反应中显示出零级动力学效应,这表明CAT与Tp*Cu的配位要比苄胺强得多。苄胺的抑制作用可以忽略不计。作者认为,Tp*配体使铜催化剂具有较软的路易斯酸性,该路易斯酸有利于与诸如硫的软碱相互作用。
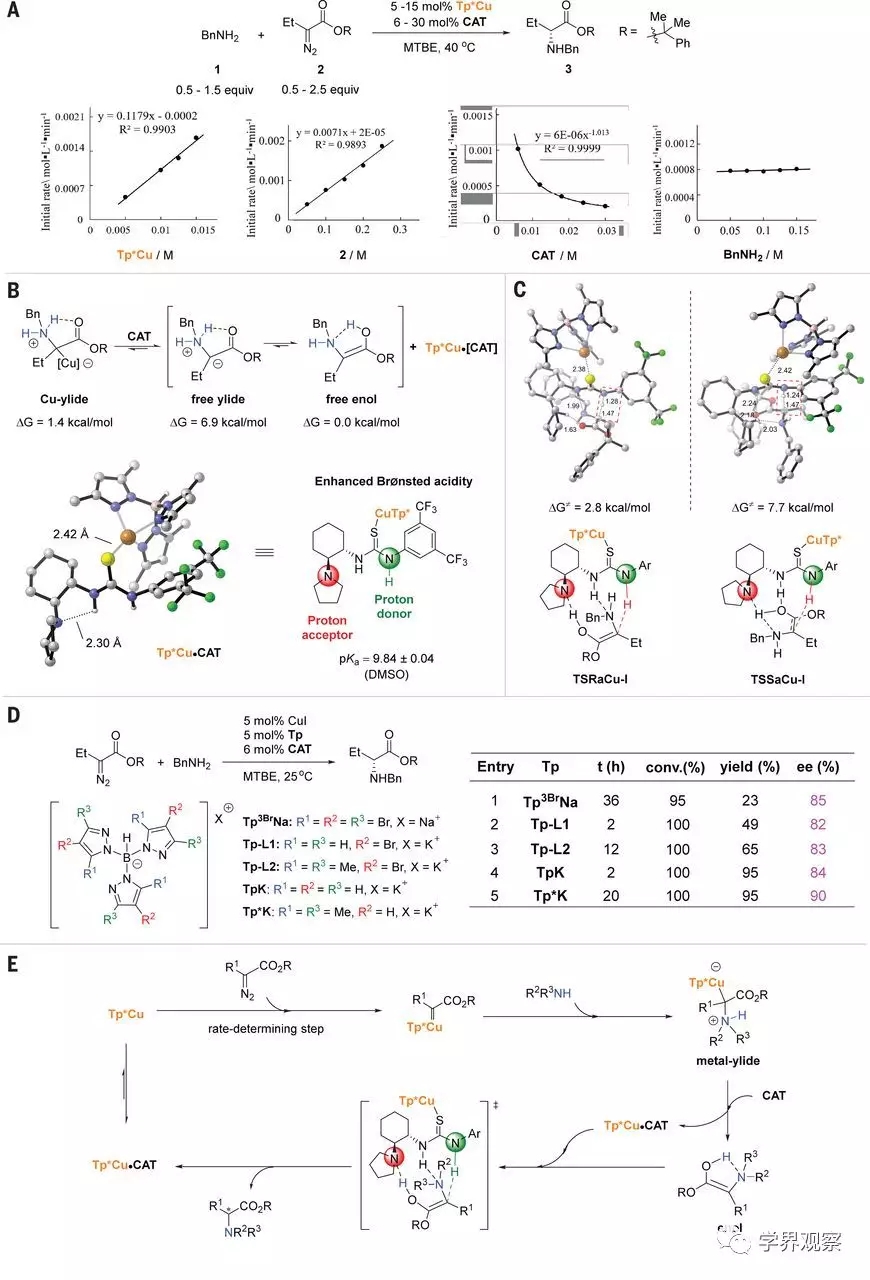
图4 机理研究。(A)N-H 插入反应的动力学曲线。(B)计算的铜-内酯,游离内酯和游离烯醇的吉布斯自由能(ΔG)。(C)DFT优化的R和S产品的最低能量跃迁结构。(D)不同Tp配体的影响。(E)推测的催化循环。
密度泛函理论(DFT)计算表明,中间体铜内酯中的铜催化剂可通过硫脲进行配位,释放出游离内酯或更稳定的互变异构体游离烯醇(图4B)。即使不加入手性催化剂,这些中间体的质子转移仍然是一个非常快速的过程,并且可以通过微量水、烯醇中间体、甚至底物本身介导。相似的pKa值促进了质子转移,其中硫脲使新形成的立体异构中心质子化,而氨基使烯醇脱质子化。使用DFT对Tp*Cu•CAT络合物进行计算揭示了一种最小能级结构,其中Tp*Cu与硫脲硫原子的键合提高了布朗斯台德酸度(图4B)。
作者以Tp*Cu•CAT配合物为催化剂,通过DFT对确定对映体的质子转移步骤进行了计算研究。图4C显示了对应于产物的主要和次要对映体的最低能量跃迁状态的结构。最佳过渡态TSRaCu-1的自由能仅比游离烯醇高2.8 kcal/mol,这意味着质子转移催化极为有效。根据实验观察,过渡态TSRaCu-I的计算能量比TSSaCu-I过渡态的计算能量低4.9 kcal / mol,从而导致了不利的(S)-NH插入。除了在过渡态TSRaCu-1和TSSaCu-1中不同的氢键相互作用之外,TSRaCu-1中的S-Cu键明显短于TSSaCu-1中的S-Cu键。S-Cu键越短,表明硫脲对铜的配位作用越强,硫脲催化剂的布朗斯台德酸度可能更高,这将促进质子转移到底物上。
在标准反应条件下,作者还评估了其他几个在吡唑环上带有不同取代基的三(吡唑基)硼酸酯(Tp)配体(图4D)。尽管收率波动很大,但原位红外研究表明,所有测试的Tp配体均促进了高转化率。当使用相同的手性硫脲催化剂时,修饰Tp配体也会影响对映选择性,表明铜催化剂参与了对映体确定步骤。相反,在调节手性硫脲催化剂的芳环的电子性质时,对映选择性急剧下降,而收率几乎保持不变。作者再次假设,铜配位可增强硫脲催化剂的布朗斯台德酸度,同时对远距离的对映体诱导位点产生最小的影响(图4C)。
基于上述机理研究,作者提出了催化循环(图4E)。Tp*Cu•CAT络合物解离释放Tp*Cu,其催化重氮酯向金属卡宾转化;脂肪族胺对金属卡宾亲核攻击产生了金属叶立德;催化剂CAT取代金属叶立德中间体中的叶立德生成游离烯醇和Tp*Cu•CAT络合物;然后,Tp*Cu•CAT复合物通过推拉机制促进游离烯醇中的质子转移:氨基部分接受来自烯醇羟基的质子,而硫脲部分将质子提供给烯醇的β-碳。
作者表示:整体转化的成功取决于非手性铜催化剂和手性有机催化剂的综合性能。这项研究不仅解决了对映选择性卡宾插入反应的长期挑战,而且为涉及强配位底物的过渡金属催化的不对称转化提供了潜在的通用策略。
哈佛大学Jacobsen在同期Science对该项研究进行了点评,表示“非手性过渡金属配合物与手性氢键供体的协同作用具有实现新的不对称转化的巨大潜力。有机过渡金属化学提供了有机催化剂无法获得的多反应模式,并且已经发现手性氢键供体催化剂可通过多种非共价机制促进对映体控制。周其林和朱守非的这项研究提供了极具说服力的验证。”

周其林院士,有机化学家。南开大学教授。1957年2月生于江苏南京。1982年7月毕业于兰州大学化学系,1985年、1987年先后获中国科学院上海有机化学研究所硕士、博士学位。先后在德国Max-Planck高分子研究所、瑞士Basel大学和美国Trinity大学从事博士后研究。2009年当选中国科学院院士。主要从事不对称催化合成方面的研究。涉及手性催化剂的结构设计、催化剂合成、相应的各种不对称合成反应研究和应用这些手性催化剂合成手-物分子等。发展了一类全新的手性螺环配体,并以该类配体为基础设计合成了一系列新型手性螺环催化剂。这些催化剂在不对称催化氢化、碳-碳键和碳-杂原子键形成等一系列不对称合成反应中都表现出优秀的催化活性和对映选择性。研究成果已经在手-物等合成中获得应用。

朱守非教授,2000年和2005年在南开大学化学学院分别获得理学学士和理学博士学位;2012-2013年在日本东京大学做博士后;2005年至今在南开大学化学学院工作,2016年获国家自然科学基金委杰出青年基金资助。长期从事催化有机合成化学研究,重点研究了几类以氢转移为关键步骤的重要有机合成反应,提出了“手性质子梭”概念,为金属催化的不对称质子转移反应提供了全新的解决方案;发现了催化卡宾对硼氢键的插入反应,为有机硼化合物的合成提供了新的方法;发展了多种用于烯烃氯化和硅氢化反应的高效催化剂,实现了多种重要生物活性分子的高效合成。迄今发表研究论文100余篇,编写著作章节2篇:获授权中国专利5项。曾获天津市自然科学一等奖2项(均为第3完成人),中国化学会青年化学奖,中国化学会青年手性化学奖,天津青年科技奖,天津青年五四奖章,Asia Core Program Lectureship Award等奖项。

第一作者南开大学李茂霖(Mao-Lin Li)
参考资料:
https://science.sciencemag.org/content/366/6468/990
https://science.sciencemag.org/content/366/6468/948

