最近几年,对于亚甲基不对称C−H键转化已经得到了比较快速的发展,对于分子内的反应已经有很多报道。相比之下,基于分子间反应将非活化的亚甲基转化为其他官能团取代的手性碳中心仍然具有很大的挑战性。在这一领域余金权课题组用其发展的单齿导向基团,设计了一系列基于氨基酸或者喹啉类的双齿手性配体,实现了亚甲基上sp3碳氢键的不对称芳基化和氟化反应等[1-2],为基于导向的不对称碳氢键活化反应的发展开辟了新的思路。
相比于单齿导向基团,双齿导向基团多占据了金属的一个配位点,因此很难寻找到合适的手性配体来实现对映选择性控制。2015年段伟良课题组报道了8-氨基喹啉(AQ)导向的钯催化的脂肪族酰胺不对称C(sp3 )−H键活化反应,该反应使用简单易得的BINOL骨架的磷酰胺或磷酸作为手性配体,能够到达82%的ee值[3]。随后,2016年南开大学陈弓、何刚团队实现了2-吡啶甲酸(PA)导向的胺类化合物的γ位C(sp3)−H键不对称芳基化反应,ee值高达97%[4]。最近,浙江大学的史炳锋课题组用其发展的PIP双齿导向基团,在6,6'-位修饰的BINOL磷酸配体的参与下实现了脂肪酰胺类化合物β位C(sp3)−H键的不对称芳基化反应,在该反应体系中作者用的是反应活性相对较低的芳基溴代物作为偶联试剂,ee值最高能达到90% [5] 。这些工作为双齿导向不对称碳氢键活化打下重要了基础。
近日,南开大学陈弓、何刚研究团队与南开大学彭谦研究员合作,成功的实现了AQ导向的零价钯(Pd(0))催化的分子间的C(sp3)−H键的不对称芳基化反应。该反应用Pd2(dba)3•CHCl3为催化剂,BINOL骨架的亚磷酰胺作为手性配体,芳基碘苯作为芳基化试剂,以高达95%的ee值实现了脂肪族酰胺β位C(sp3)−H键的不对称芳基化反应。
首先,作者对反应的催化剂和配体进行了考察(图1),发现当使用Pd(II)作为催化剂与P(III)作为手性配体时,反应的ee值要比Pd(II)与P(V)结合时略高(entry 4)。因此,作者想如果直接用Pd(0)与P(III)共同作用是否能实现更高的对映选择性控制。于是,作者又考察了一系列的Pd(0)催化剂和P(III)配体,发现在10 mol% 的Pd2(dba)3•CHCl3为催化剂,30 mol%的BINOL骨架的亚磷酰胺(L4)作为手性配体时,能够达到91%的ee值(entry 16)。随后作者发现随着芳基碘苯的量的增加反应的ee值也随之增加,另外额外加入30 mol%的dba-3-CF3作为添加剂,不仅能够提高反应的收率,而且也能略微提高反应的对映选择性(entry 21)。
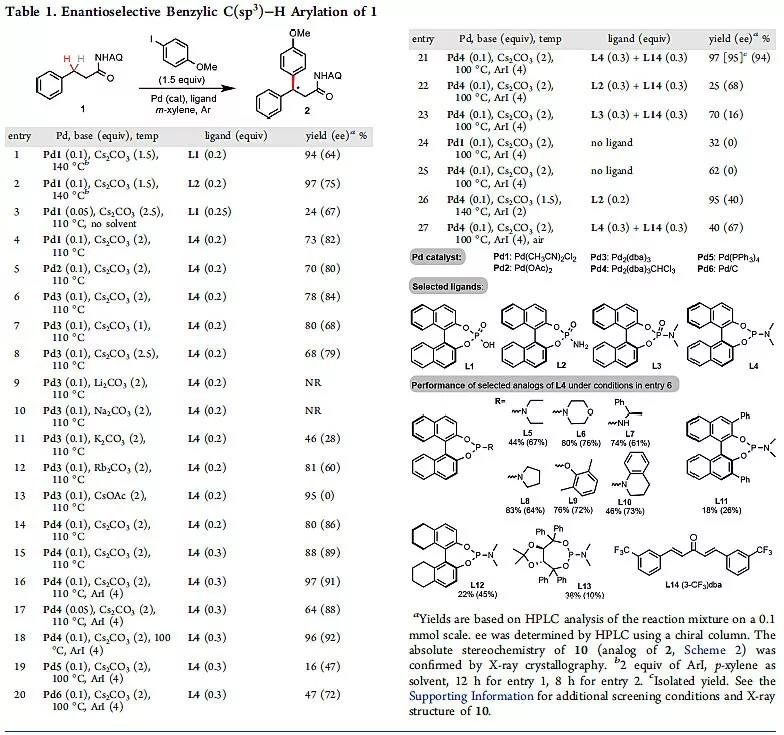
图1. 条件筛选
确定了最佳反应条件后,接下来就是对底物的扩展,选用化合物1作为模板底物,对芳基碘代物进行筛选(图2)。结果显示,芳基碘上可以带有卤素、酯基、三氟甲基、烷基、烷氧基等,且都取得了良好的收率和良好的选择性,而且从表中可以看出,随着取代基吸电子性的增强,反应的收率也随之下降,这在后面的机理研究部分会做详细介绍。
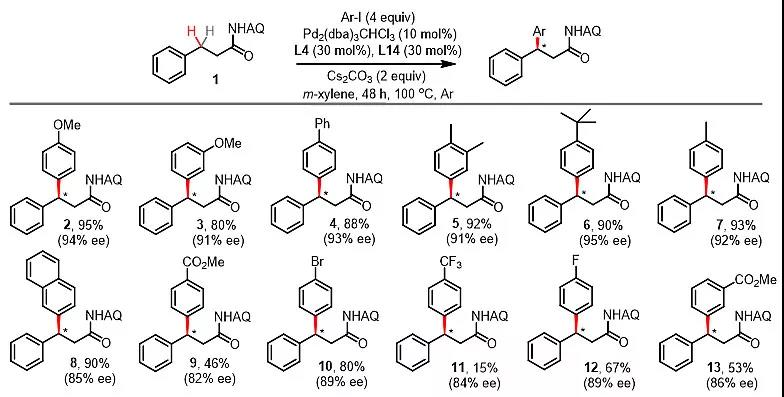
图2. 芳基碘苯取代基的考察
除了对碘代物进行扩展以外,作者还对酰胺类底物进行了扩展,如图所示(图3),底物中的R基团为芳基时,如果芳基上的取代基在间位时,能够实现较高的收率和对映选择性;如果R基团为烷基链时,反应性和对映选择性都比较差,这也是该反应的不足之处。
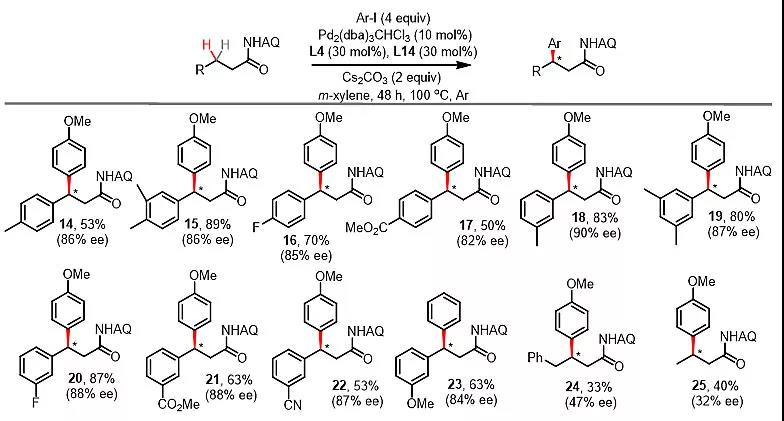
图3. 酰胺类底物的考察
在得到以上的实验结果后,作者对反应的机理进行了详细的研究。KIE实验表明,当芳基碘苯的电性不同时,KIE实验呈现出不同的实验结果(图4),当芳基碘苯含有吸电子取代基时,没有呈现出明显的KIE效应,也就是说在吸电子碘苯参与反应时,反应的决速步不是碳氢键活化,可能是还原消除(计算结果表明氧化加成这一步所需的能量很低)。
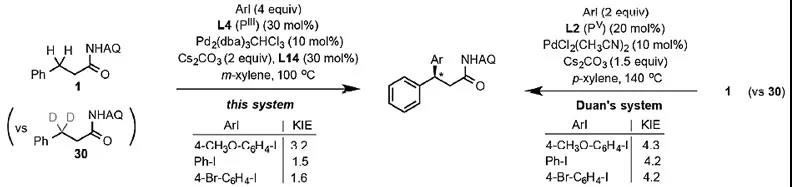
图4. KIE实验
在对芳基碘苯上的取代基进行考察时,作者就发现芳基碘苯的电性对反应的活性有比较明显的影响。哈米特方程曲线(图5)与实验结果一致,即当取代基的吸电子性增强时,反应速率逐渐降低。作者推测,在反应的决速步骤中可能有芳基碘苯的参与,这又进一步说明芳基碘苯参与的还原消除这一步是反应的决速步。
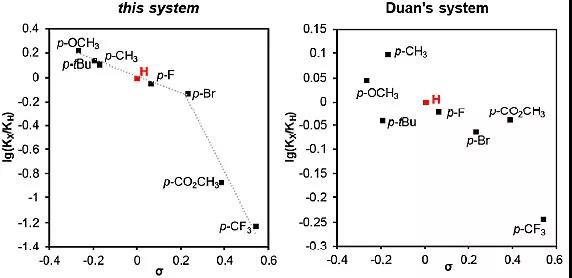
图5. 哈米特方程曲线
随后,作者对反应的动力学方程进行了研究(图6),发现该反应体系的速率对底物是零级的,对催化剂是一级的,对芳基碘苯也是一级的并且有动力学饱和现象。作者对段伟良的反应体系的动力学方程也进行了研究,发现在他们的体系中反应速率对底物和催化剂是一级的,对芳基碘苯是零级的。这就表明这两种反应体系经历了不同的反应历程。在本文的反应体系中还原消除是决速步,而段伟良的体系中碳氢键活化是决速步。

图6. 哈米特方程曲线
DFT计算表明,BINOL亚磷酰胺配体和碳酸铯碱都参与了不对称C(sp3)−H键活化的立体选择性控制,并且C-H钯化和还原消除需要的能量很接近。根据以上实验结果和机理研究,作者提出了一个可能的反应机理,如图7所示。
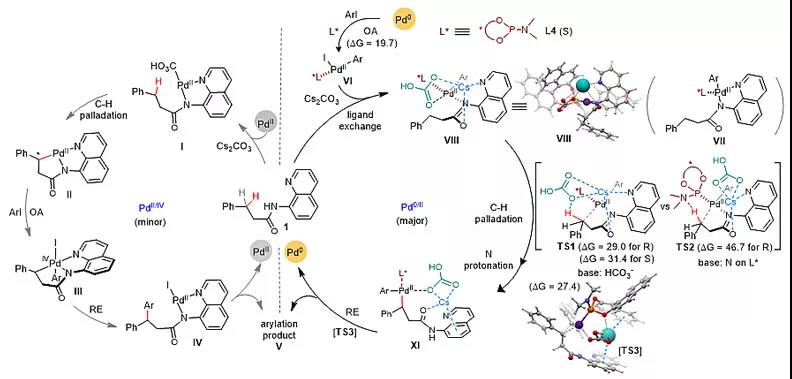
图7. 可能的反应机理
总的来说,与Pd(II)/Pd(IV)催化循环的双齿导向碳氢键活化相比,本文用Pd(0)作为催化剂,通过Pd(0)/Pd(II)催化循环首次用双齿导向基团辅助实现了分子间的不对称C(sp3)−H键芳基化反应。相关工作最近发表在ACS Catalysis 上,通讯作者为南开大学的陈弓教授、何刚研究员和彭谦研究员。南开大学的博士生童华绒是该论文的第一作者,硕士生郑素娟负责完成其中的计算化学工作。
该论文作者为:Hua-Rong Tong, Sujuan Zheng, Xinghua Li, Zhiqiang Deng, Hao Wang, Gang He, Qian Peng, and Gong Chen.
原文:
Pd(0)-Catalyzed Bidentate Auxiliary Directed Enantioselective Benzylic C−H Arylation of 3‑Arylpropanamides Using the BINOL Phosphoramidite Ligand.
ACS Catal., 2018, 8, 11502−11512, DOI: 10.1021/acscatal.8b03654.
参考文献
1. Ligand-accelerated enantioselective methylene C(sp3)–H bond activation. Science 2016, 353, 1023-1027. doi:10.1126/science.aaf4434.
2. Controlling Pd(IV) reductive elimination pathways enables Pd(II)-catalysed enantioselective C(sp3)−H fluorination. Nat. Chem., 2018, 10, 755-762. doi: 10.1038/s41557-018-0048-1.
3. Palladium-Catalyzed Asymmetric Arylation of C(sp3)−H Bonds of Aliphatic Amides: Controlling Enantioselectivity Using Chiral Phosphoric Amides/Acids. Org. Lett., 2015, 17, 2458-2461. doi:10.1021/acs.orglett.5b00968.
4. An Enantioselective Bidentate Auxiliary Directed Palladium-Catalyzed Benzylic C-H Arylation of Amines Using a BINOL Phosphate Ligand. Angew. Chem. Int. Ed., 2016, 55, 15387 –15391. doi:10.1002/anie.201609337.
5. Yan, S. -Y.; Han, Y.-Q.; Yao, Q.-J.; Nie, X.-L.; Liu, L.; Shi, B.-F., Palladium(II)‐Catalyzed Enantioselective Arylation of Unbiased Methylene C(sp3)−H Bonds Enabled by a 2‐Pyridinylisopropyl Auxiliary and Chiral Phosphoric Acids. Angew. Chem. Int. Ed., 2018, 57, 9093 –9097. doi:10.1002/anie.201804197.

