过渡金属催化的环丙烷与π-不饱和化合物的对映选择性环加成,是构建手性五元环状结构最为重要的方法。然而,由于环丙烷C–C键能较高,活化困难,已有的方法主要局限于含高环张力或强吸电子基团的环丙烷(图1a, Type I),难以应用于价廉易得且易于转化的环丙基酮类弱活化环丙烷(图1a, Type II)。高对映选择性环丙基酮的环加成反应的关键难点在于:酮羰基的亲电性较强,在C–C键断裂所需的高温或路易斯酸条件下,产物的手性α-叔碳中心难以稳定存在,容易发生外消旋化(图1b)。因此,自2006年以来,尽管环丙基酮消旋的环加成反应已有成功发展,但是高对映选择性版本一直难以实现。为了解决这一难题,一些底物修饰的方法被发展起来。例如,将酮替换为亲电性较弱的酰胺,可以在130 °C下,实现与炔烃的高对映选择性环加成(图1b左)。或者采用多取代环丙烷来稳定自由基中间体,发展温和条件下自由基型环加成反应(图1b右)。尽管这些方法可以实现高对映选择性的环加成反应,但是底物结构的特殊需求显著限制了产物的结构的多样性,导致反应的应用较为局限。因此,如何通过催化剂的调控,实现简单环丙基酮的高效不对称环加成反应是当前该领域亟待突破的关键挑战。
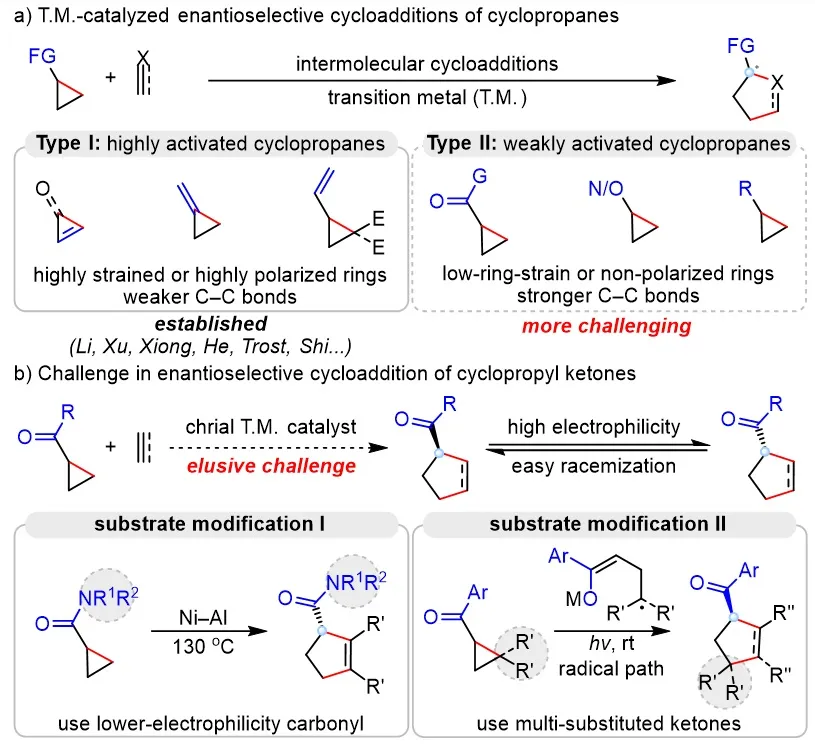
图1. 研究背景
最近,南开大学叶萌春/徐魏魏团队提出了一种创新的催化策略:通过设计新型手性二胺-膦氧(PO)配体,获得了该类环加成反应活性的显著提升,可以使反应即使在室温下都可以高效进行,从而有效减弱了产物的消旋化。此外,引入弱配位的四氢呋喃(THF)溶剂,进一步降低Al中心的Lewis酸性,抑制产物的消旋化。最终,在温和条件下(室温),顺利实现了环丙基酮和炔烃的高对映选择性[3+2]环加成反应,不仅具有优异的底物普适性,而且能获得高达99%的收率和99%的对映选择性(图2)。机理研究表明,其高反应活性与立体选择性主要源于三个关键因素:新型PO配体的高活性,经PO/THF协同调控后适度减弱的Lewis酸性,以及温和的反应温度。这些因素的协同作用有效抑制了烯醇化介导的消旋路径,同时避免了酮羰基高亲电性引发的副反应。

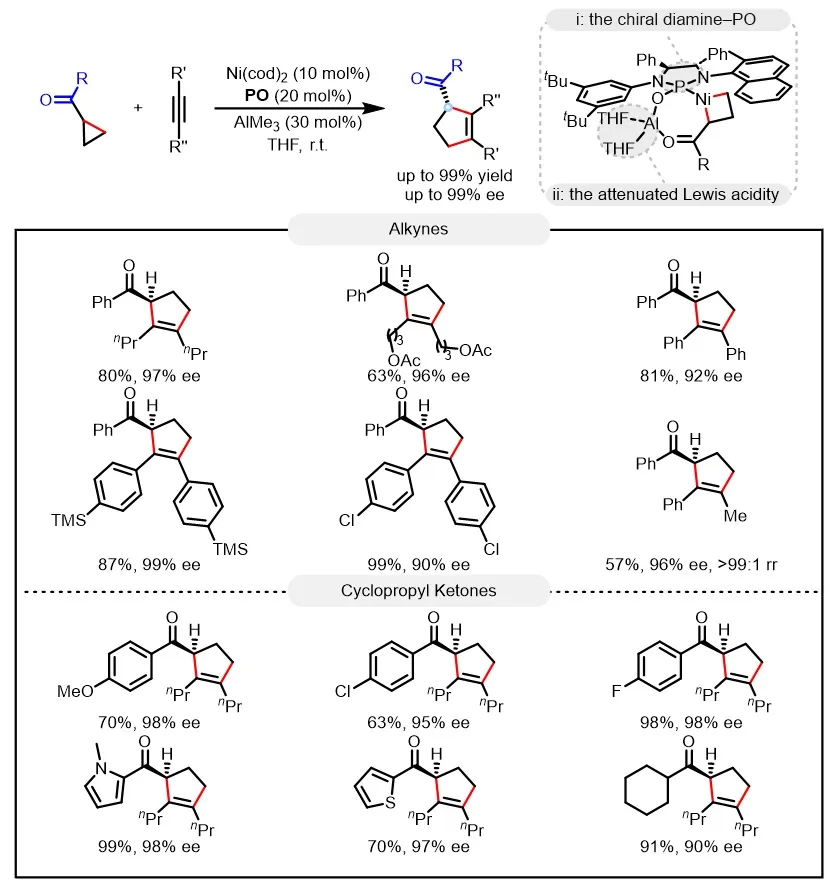
图2. 镍铝双金属催化环丙基酮的不对称环加成反应
为深入验证该机理,该团队开展了一系列实验。首先考察了手性产物在不同温度下的稳定性:产物在室温下构型稳定,但随温度升高逐渐消旋(图3a)。进一步研究发现,在该催化体系中,无论使用手性或消旋PO配体,反应均专一地生成构型保持的产物(图3b)。这一结果与以往体系中常观测到构型翻转的现象形成鲜明对比,证实了PO/THF协同调控对Lewis酸性的削弱作用,以及该体系在温和条件下对中间体的稳定能力,从而同步实现了高活性与高对映选择性。
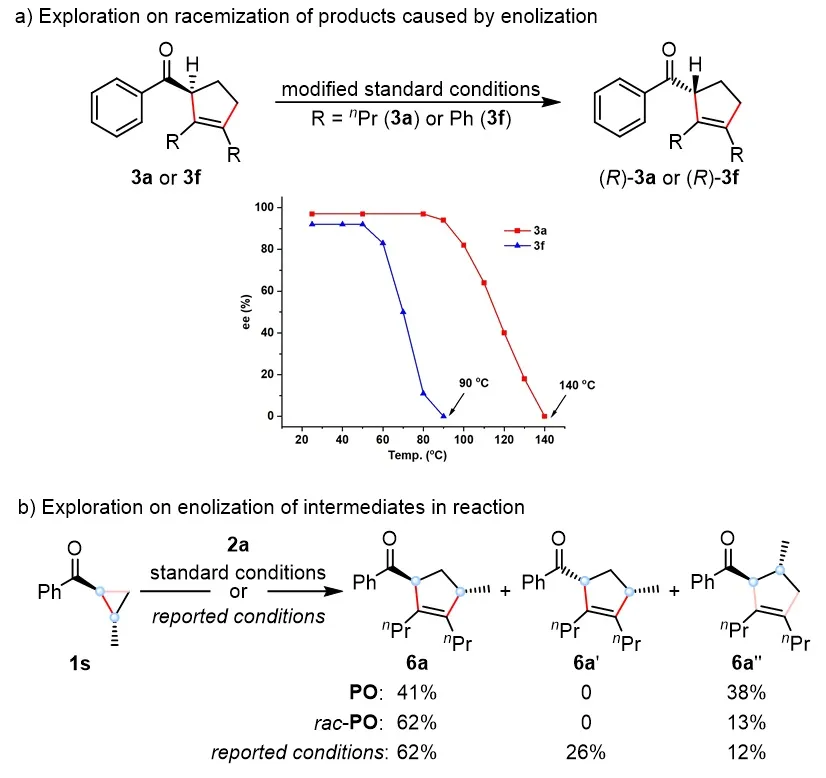
图3. 机理研究实验与反应机理
总而言之,叶萌春/徐魏魏团队开发了一种高效手性PO配位的镍-铝催化剂体系,能够在温和条件下实现简单环丙基酮与炔烃的对映选择性环加成反应,能以优异的收率(高达99%)和对映选择性(高达99% ee)高效合成具有α-叔碳手性中心的环戊基酮。其高选择性得益于配体与路易斯酸的协同作用,有效抑制了烯醇化和副反应的发生。该反应具有广泛的官能团耐受性,并已成功应用于生物活性分子的衍生化,凸显了其在利用简单前体快速构建多样化药物分子骨架方面的应用价值。
原文(扫描或长按二维码,识别后直达原文页面):

Enantioselective Ni–Al Bimetal-Catalyzed Cycloaddition of Cyclopropyl Ketones with Alkynes under Mild Conditions
Yinpeng Wang, Yi Li, Haorui Wang, Lujie Ji, Yu Fu, Xingzhong Gao, Qi-Lin Zhou, Weiwei Xu*, Mengchun Ye*
J. Am. Chem. Soc. 2025, DOI: 10.1021/jacs.5c18087

