对映选择性过渡金属催化烯烃的羰基化环化反应是合成含有手性α-季碳的γ-内酰胺的一种最有吸引力的方法之一。目前报道的反应往往需要使用活性较高的C(O)-Cl/F作为羰基前体,而以惰性C(O)-H键为羰基前体的环化反应尚未实现,尽管其稳定性更好。原因主要是惰性C-H键的活化往往需要更苛刻的条件但却会加速不稳定羰基金属间体的分解。为解决这一问题,南开大学叶萌春课题组使用连萘胺衍生的膦氧为手性配体,通过镍和铝双金属催化以高对映选择性和区域选择性实现了C(O)-H键与烯烃的羰基化环化反应。在该反应中,原料中大位阻的N-保护基和膦氧配体中柔性的N-烷基取代基对于反应活性和选择性至关重要。最终该反应以高达99%的收率和高达98%的ee构建了一系列含有手性α-季碳的γ-内酰胺。
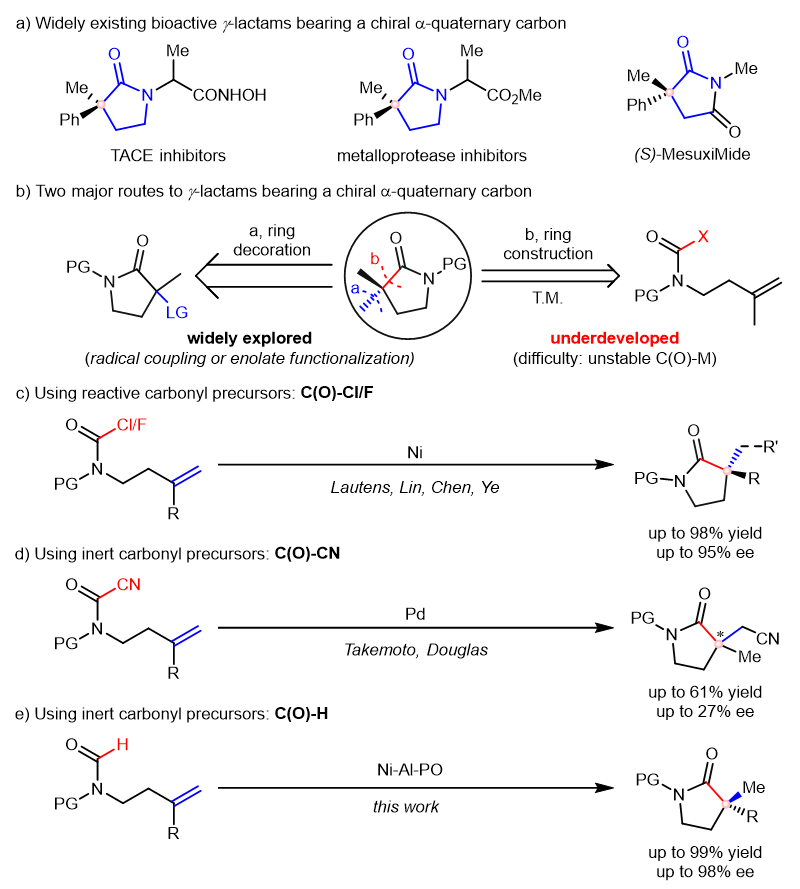
含有手性α-季碳的γ-内酰胺是各种生物活性分子和药物分子中的核心结构单元。然而,这些γ-内酰胺的合成始终是一个巨大的挑战,因为以高立体选择性构建含有羰基的季碳非常困难。为解决这一问题,直接修饰γ-内酰胺的策略被开发出来,而在这种方法中,消旋的γ-内酰胺需要预先制备出来,随后通过自由基偶联或烯醇官能化等立体选择性转化引入手性季碳;为简化这一步骤,一步环化的策略应运而生,其中,连有烯烃的合适的羰基前体在过渡金属催化的条件下发生羰基化环化反应,在此过程中,环状的γ-内酰胺和手性羰基季碳同时被构建出来。尽管一步环化的策略具有原料价廉易得且步骤经济等优势,但目前这种策略始终面临着羰基金属中间体在环化前容易分解等问题。为此,活性较高的羰基前体常常被用来促进环化反应,如C(O)-Cl/F等。然而,这种模式却始终无法实现更为惰性的羰基前体如C(O)-C/H参与的环化反应,可能是因为:(1)惰性C(O)-C/H键的活化往往需要相对剧烈的条件,而在此条件下不稳定的羰基金属中间体更易在环化前分解;(2)分解产生的一氧化碳具有较强的配位能力,这不仅抑制了金属的反应性而且阻止了手性配体与金属的配位,从而导致反应的活性和选择性都下降。如,以C(O)-C为羰基前体时,反应收率最高只有61%而ee值最高只有27%,而且以C(O)-H为羰基前体构建含有手性α-季碳的γ-内酰胺则从未实现。为了克服这一挑战,南开大学叶萌春课题组使用连萘胺衍生的膦氧为手性配体,通过镍和铝双金属催化以高对映选择性和区域选择性实现了C(O)-H键与烯烃的羰基化环化反应。在该反应中,底物上大位阻的N-保护基和膦氧配体中柔性的N-烷基取代基对于反应活性和选择性至关重要。最终该反应以高达99%的收率和高达98%的ee构建了一系列含有手性α-季碳的γ-内酰胺产物,这些产物要么本身是生物活性分子,要么是合成活性分子的关键前体。
叶萌春课题组使用连萘胺衍生的膦氧配体、镍和铝双金属催化剂合成了含有季碳手性中心的吲哚酮。然而,在这项工作中,对于原料而言,羰基和烯烃中间的连接链必须是刚性的芳环,其它非芳环的底物则没有活性,可能是因为惰性C(O)-H键难以活化且C(O)-M中间体难以环化。为解决这一问题,他们假设在酰胺上引入大位阻的N-保护基能够将柔性链推向金属镍催化中心,此外,在连萘胺衍生的膦氧上引入柔性链保护基或许能够兼容原料中的烯烃单元,进而提高反应活性和选择性。

基于上述假设,作者在以往报道的最优条件的基础上对反应条件进行了系统的考察,结果表明甲酰胺上大位阻的N-保护基对反应活性至关重要:位阻小的甲基、乙基、苄基和取代的苄基都没有活性,只有大位阻的2,4,6-三甲基苄基(N-BnTM)是最优的保护基,能够以21%的收率、68%的ee值和高达>99:1的区域选择性构建目标的γ-内酰胺产物。随后,以N-BnTM甲酰胺为模板底物,考察了一系列手性连萘胺衍生的膦氧配体,最终他们通过替换芳基为烷基取代基,将反应的收率提升至94%,ee值提升至87%。在最优保护基和最优配体的基础上,他们进一步考察了其它的反应条件:在筛选的路易斯酸中,三甲基铝仍然是最优条件,当其用量提升至40%以后,反应的收率提升至99%、ee值提升至94%而区域选择性保持;而其它的路易斯酸通常会降低反应的收率或者区域选择性。另外,温度的考察表明,当温度降至80 ℃,反应的ee值会进一步提升至97%。

在最优条件的基础上,作者对底物的适用范围进行了考察。首先,作者考察了烯烃上的取代基对反应的影响,结果表明:含有给电子基的芳环都能很好地被反应兼容,如甲氧基(2)、甲基(3)、乙烯基(4)和硅基(5)。基于产物5的单晶结构,手性季碳的绝对构型被定为S。类似地,反应也能兼容各种吸电子取代基,如氯(6)、氟(7、8)和三氟甲基(10),以60%-99%的收率和94%-97%的ee值构建目标产物。但邻氟芳基取代的化合物参与反应时,反应的收率降低,仅为32%,但反应的ee较高,为98%。作者认为,邻位取代基会导致芳基旋转,进而影响烯烃与配体的相互作用。此外,反应也能对芳杂环兼容,如二苯并呋喃(11,92%,93% ee)和呋喃(12, 62%,81%ee)。
在最优配体的条件下,作者还进一步考察了甲酰胺底物中氮原子上的保护基对反应的影响。实验结果表明反应除了不能兼容直链烷基取代基外,其它各种环烷基(13-15)和支链烷基(16)都能被很好地兼容,反应收率为75%-99%,ee值为90%-96%。另外,各种芳基保护基也能被兼容,如,简单的苯基(17,97%,98% ee)、含有给电子基的芳基(18-22, 51%-85%, 94%-98% ee)、含有吸电子基的芳基(23-26, 65%-90%,92%-98% ee)。需要注意的是,反应对大位阻的邻位取代基也能兼容(22),但是反应对二烷基或三取代的烯烃不兼容,同时,缩短或延长烯烃链长也会导致环化反应失败。

为了证明当前反应的实用性,作者进行了相应的产物转化并合成了生物活性分子。首先,作者进行了模板反应的克量级实验并顺利地得到了目标产物1;虽然反应收率略有降低,但反应的ee值仍然保持。其次,在氢化铝锂的条件下,γ-内酰胺能够以80%的收率和97%ee值转化成相应的吡咯烷。另外,在酸性条件下,2,4,6-三甲基苄基保护基能够很容易被脱除并生成相应的化合物28,而28是很多生物活性分子的关键合成前体,如28能够转化成含有手性季碳的氨基酸或者抗心律失常的化合物30等。值得注意的是,28能够被氧化成具有生物活性的琥珀酰亚胺31,而31同样是多种生物活性分子的关键合成前体,如31能够转化成抗惊厥药甲琥胺32、具有镇痛作用的化合物33、非竞争性mGluR2拮抗剂34等。

为了探究反应机理,作者进行了相关的机理实验。氘标记实验表明,甲酰胺的H完全转移到烯烃末端。在平行实验中没有观察到明显的动力学同位素效应(kH/kD=1.00),这表明甲酰胺C-H键的活化没有参与决速步。同时,作者还进行了非线性效应研究,发现产物的ee值和手性膦氧配体的ee值是线性相关的,因此作者认为在决定产物对映选择性的过渡态中只涉及了一分子手性膦氧配体。此外,作者与天津大学黄跟平团队合作通过DFT计算对反应的机理进行了深入的研究,结果表明:S1和PO-Ni-Al催化剂配位生成中间体IM1,在此基础上,产物1经由氧化加成、迁移插入、还原消除生成。其中,C-C键还原消除是反应的绝速步,这和KIE实验结果一致;此外,该反应的迁移插入是不可逆的,这与氘标记实验结果一致,同时,反应的对映选择性也由迁移插入这一步决定。DFT计算还表明,副产物1’可能是经由LLHT、还原消除的机理生成的,而LLHT比C-H键氧化加成的能量更高,这解释了为什么反应的区域选择性高达>99:1。

总结与展望
南开大学叶萌春课题组以高区域选择性和对映选择性实现了甲酰胺C-H键和烯烃的羰基化环化反应,最终以高达99%的收率和高达98%的ee值构建了一系列含有手性α-季碳的γ-内酰胺产物,而这些γ-内酰胺产物是很多生物活性分子的关键合成前体。在该反应中,原料中大位阻的N-保护基和膦氧配体中柔性的N-烷基取代基对于反应活性和选择性至关重要。
相关研究成果发表于Journal of the American Chemical Society。南开大学化学学院元素有机化学国家重点实验室的博士研究生徐魏魏为该文第一作者,天津大学黄跟平教授和南开大学化学学院元素有机化学国家重点实验室叶萌春教授为该工作的通讯作者。相关工作得到国家重点研发计划、国家自然科学基金委等项目的支持。
原文(扫描或长按二维码,识别后直达原文页面):
Enantioselective Carbonylative Cyclization of Alkenes with C–H Bonds for Synthesis of γ-Lactams Bearing an α-Quaternary Carbon
Weiwei Xu, Yanan Sun, Yuqing Jiang, Xueyuan Yan, Zhixuan Gao, Haorui Wang, Genping Huang*, Qi-Lin Zhou, Mengchun Ye*
J. Am. Chem. Soc., 2024, DOI: 10.1021/jacs.4c15875

