酶催化的糖苷化是自然界修饰生物活性分子的重要手段,许多带有糖基化修饰的天然产物都表现出优异的生物活性。值得注意的是除了常见的O-糖苷和N-糖苷以外,自然界还通过酶催化的碳氢键官能团化策略制备了许多结构新颖、功能特异的芳基糖苷类产物。由于芳基糖苷结构比O-糖苷和N-糖苷具有更高的稳定性和更好的成药性,碳苷化也是糖缀药物改造的有效策略。例如近年来上市的一系列治疗二型糖尿病的SGLT2抑制剂,包括达格列净 (dapagliflozin)、卡格列净和依帕列净等等都是由O-糖苷先导化合物衍变而来。因此,芳基糖苷类化合物的高效合成在糖化学和医药研发领域受到越来越多的重视。
传统的芳基糖苷合成方法分主要有以下几种:一是利用富电子芳烃作为偶联组分,在当量Lewis酸存在下与糖给体发生傅克类型的糖基化反应;另一种是利用预官能团化的芳基金属试剂(如芳基锂、芳基锌、芳基镁试剂等)与糖基给体进行偶联构筑C-糖苷键。这些方法往往存在着区域选择性和立体选择性差、底物适用范围窄,以及芳基金属物种预官能团化操作繁琐等局限。因此发展高效、高立体选择性的芳基糖苷合成方法对于对糖化学领域和医药研发都具有重要的意义。
受到自然界酶催化芳基糖苷合成策略的启发,最近南开大学陈弓-何刚团队利用钯催化的碳氢键糖基化反应实现了芳基糖苷的高效合成。该方法利用Pd(OAc)2作为催化剂,8-氨基喹啉(AQ)做导向基团,首先通过碳氢活化生成环钯中间体。随后环钯中间体作为亲核试剂与氯代糖生成的糖基氧鎓离子反应,实现芳烃(芳杂环)C(sp2)-H高立体选择性糖基化。该方法可以广泛应用于各种芳环和芳杂环底物,同时对多种氯代单糖以及寡糖适用,可以简化各种复杂芳基糖苷的合成,为药物分子的后期修饰提供了新的工具。
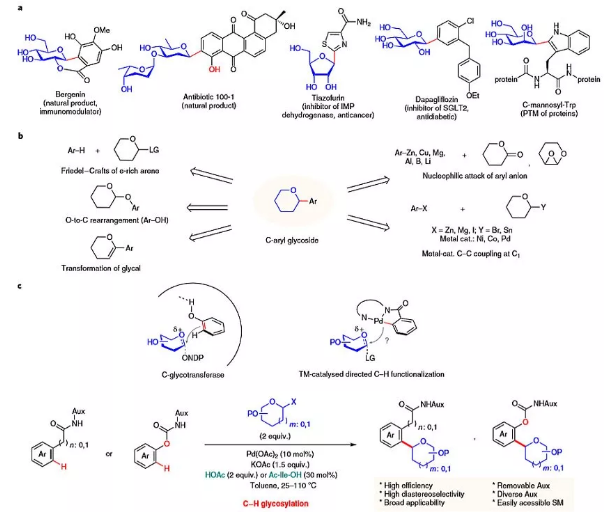
图1. C-芳基糖苷及其合成策略。图片来源:Nat. Catal.
如图1c所示,自然界使用形式上的C-H键官能团化策略来催化C-芳基糖苷的生物合成。虽然在Lewis酸条件下,富电子芳烃的C-糖基化反应采用了类似的傅克型机理,但缺乏区域控制限制了其在简单底物上的应用。使用化学计量的金属芳烃作为σ亲核试剂进行糖基化,能进行精确的区域控制。尽管如此,除了需要的预官能团化步骤外,强碱性的阴离子芳基亲核试剂与最常用的糖基氧鎓正离子不相容,从而限制了芳烃和糖基团上官能团的范围。在原则上,后过渡金属催化的C-H键官能团化可以通过利用较软的芳基金属配合物中间体来避免硬芳基金属亲核试剂的反应性和相容性问题。最近,作者和其他人已经证明,在Pd催化下,带有酰胺连接的8-氨基喹啉(AQ)辅基(Daugulis首次引入)的苯甲酰胺可以进行邻位选择性C-H键官能团化。这些反应首先进行邻位导向的C-H键钯化,形成五元二价环钯中间体。值得注意的是,AQ基团的独特的N,N-双齿结合模式可以极大地增加二价钯中心的电子密度,允许其通过高价Pd中间体与亲电试剂反应。该反应体系C(sp3)中心的亲电试剂的独特反应性,促使作者研究合适的糖基供体是否能够以非对映选择性方式得到C-芳基糖苷。
如图2所示,作者发现模型底物AQ-偶联的3-甲基苯甲酰胺与2当量四苄基保护的氯代α-甘露糖(1)在1.5当量KOAc存在下110 °C的甲苯中反应,能得到C-H键甘露糖化产物2a,产率91%,具有专一的α-选择性。根据1H NMR分析,2a采用1C4构象(1C4是描述糖构型术语,吡喃糖椅式构型,碳-1位异头碳为面上,碳-4位为面下;相对应的是椅式4C1),芳基位于直立键位置。各种羧酸添加剂可以使该反应在较低的温度下进行。值得注意的是,在存在2当量HOAc的情况下反应,45 °C条件下产物2a的收率95%。加入催化量(30 mol%)的N-氨基甲酸酯或酰基保护的α-氨基酸可在45-60 °C下实现优异的转化。N-乙酰基保护的异亮氨酸(Ac-Ile-OH)为优选酸。使用溴代甘露糖得到类似的收率(93%),而相应的三氯乙酰亚胺供体在相同条件下的收率极低(10%)。
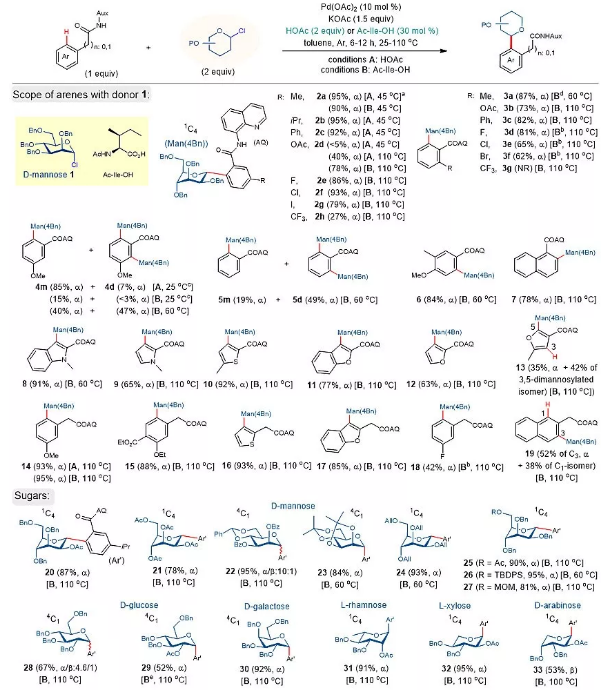
图2. 条件优化及底物扩展。图片来源:Nat. Catal.
在优化的条件下,AQ偶联的苯甲酰胺与1发生邻位C-H甘露糖基化反应,反应条件为HOAc(条件A)或Ac-Ile-OH(条件B)(图2)。一般来说,富电子芳烃在较低的温度下与HOAc添加剂反应更好。以3-甲氧基苯甲酰胺为例,在25 °C下,用2当量的HOAc反应48 h,4m产率为85%,同时得到7%的双甘露糖化物4d。在60 °C下,用30 mol%的Ac-Ile-OH得到40%的4m和47%的4d的混合物。缺电子底物的活性较低,在高温下使用Ac-Ile-OH添加剂效果较好。例如,在110 °C的B条件下,2d的产率为78%,而用HOAc的收率为40%。邻位取代苯甲酰胺需要更加剧烈的条件,在高温下反应(例如2c vs 3c)。具有相同邻位C-H键的底物通常得到单、双甘露糖基化的混合物(例如5m和5d)。杂芳烃如吲哚(8)、吡咯(9)、噻吩(10)和呋喃(11-13)的反应得到了具有良好的区域和非对映选择性的邻甘露糖基化产物。除苯甲酰胺外,2-芳基和杂芳基酰胺(14-19)还在标准条件A或B下,通过六元钯环中间体进行了邻C-H键甘露糖基化反应。
氯代糖是用于糖基化反应高效、方便的供体。如20-27所示,在标准条件下,含其他保护基的氯代甘露糖也能很好地容忍,除22以外,C-芳基甘露糖类化合物在α-选择性中形成了10:1的α/β混合物。乙酸酯(20)、4,6-苄基(22)、异丙基(23)、烯丙基(24)、叔丁基二苯基硅基(26)和甲氧甲基(MOM,27)等官能团对甘露糖具有较好的耐受性。除甘露糖外,D-半乳糖(30)、L-鼠李糖(31)、L-木糖(32)和D-阿拉伯糖(33)等其他吡喃糖的氯供体均能获得理想的C-芳基糖苷。值得注意的是,C-葡萄糖苷28是一种4.6:1的α/β混合物。2-OAc保护的葡萄糖氯供体的反应使产物29具有专一的α-选择性,表明相邻基团的参与对该反应的非对映选择性影响不大。
除AQ外,其他酰胺连接的双齿辅基也可在标准条件下以不同的效率促进邻C-H键加成反应。AQ基上的5-甲氧基取代基(MQ)不改变反应活性(2a-i)。N-甲基化AQ辅基不产生C-糖基化产物(2a-ii),而2-(甲基硫基)苯胺(SA)辅基的产物(2a-iii)产率较高。普通苯胺辅基的底物没有产生任何想要的C-糖基化产物。
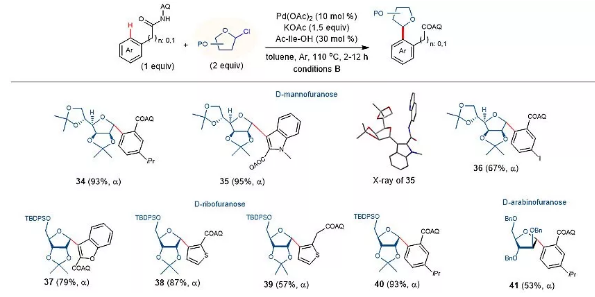
图3. C-芳基呋喃糖苷的合成。图片来源:Nat. Catal.
含有芳杂环的呋喃糖碳苷作为核苷类似物在药物开发,特别是在病毒学和肿瘤学领域中得到了广泛的应用。如何高效、高立体选择性地合成这种骨架仍然具有挑战性。如图3所示,AQ导向的碳氢键糖基化策略可以顺利实现该类芳基糖苷的构筑。例如,芳基底物与2,3:5,6-二O-异丙基-甘露糖基给体反应,可以以>90%的收率和专一的立体选择性得到产物34和35。35的结构经x射线单晶衍射证实。苯并呋喃、噻吩酰胺、2-噻吩乙酰胺与5-TBDPS-2,3-O-异丙基氯代D-核糖的反应均具有较高的收率和专一的立体选择性(37-40)。苄基保护的氯代阿拉伯呋喃糖也能以专一的α立体选择性生成产物41。
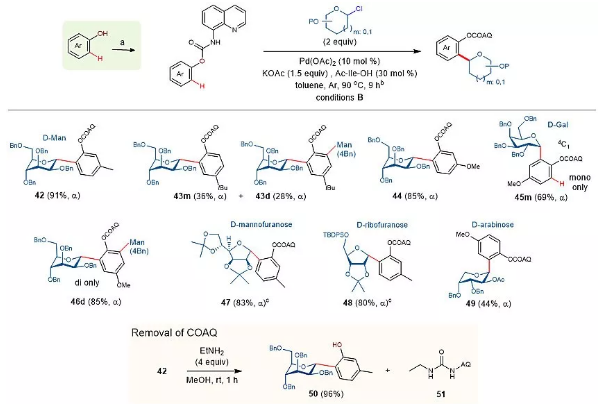
图4. 氨基甲酸酯连接的酚类底物的糖基化反应。图片来源:Nat. Catal.
苯酚邻位的C-糖苷是天然产物中最常见的芳基C-糖苷(见图1a)。这种结构的制备通常依赖于以下两种方法:第一种是通过保护的苯酚与糖给体发生傅克反应;另外一种是通过在路易斯酸促进条件下酚类O-糖苷发生分子内重排反应生成相应的芳基C-糖苷。这两种方法同样具有底物适用范围窄和反应选择性差的缺点。如图4所示,通过三光气氯化反应和酰胺缩合反应,可以将甲酸酯连接的AQ基团安装到苯酚底物上。这类底物可以与吡喃氯代糖、呋喃氯代糖在标准条件反应,以高收率和立体选择性得到预期的C-糖苷化产物。此外,作者发现在室温条件下将碳苷产物与乙胺反应,可以以近乎定量的分离收率得到游离酚产物50。值得注意的是,该反应是首次将氨基甲酸酯连接的AQ辅助基团,用于金属催化的碳氢键官能团化反应。
这种Pd-催化的AQ导向的C-H糖基化在标准条件下可以很容易地应用于更复杂的底物上(图5a)。一种保护的四糖能够以专一α-选择性的C-甘露糖化反应连接到3-异丙基苯甲酰胺的邻位上(52)。对于3-甲氧基-4-甲基苯甲酰胺,通过C-葡糖键,以95%的产率和9:1的α/β-选择性将保护的三糖麦芽糖连接在其C6位上(53)。AQ偶联抗糖尿病药物雷格列奈(含游离二级酰胺)能得到54,具有专一的α-选择性,为药物分子的后期修饰提供了一种新的策略。如图5b所示,酰胺连接的小位阻苯甲酰胺在温和的条件下,通过两步的Boc活化和LiO2H裂解得到苯甲酸的产物(56)。另外,用Schwartz试剂Cp2ZrHCl(3当量)选择性地还原2a的酰胺键,得到苄醇59。用1.5当量Cp2ZrHCl还原酰胺,选择性地生成苯甲醛产物60。C-芳基甘露苷57含有2-OAc基团,用4当量K2CO3在45 °C的湿MeOH中反应,可以得到58%的苯甲酸产物。二氯甲烷中用EDCI处理苯甲酸58,两步反应得到六元内酯62,产率85%。58的2-羟基可先被释放,然后分子内进攻酰胺基形成内酯62,然后在原位水解得到58。类似的C-芳基葡萄糖苷、半乳糖苷、鼠李糖苷和阿拉伯糖苷在类似条件下的反应,得到内酯产物61和63-65,产率优良。基于这种反应,能够快速获得天然产物bergenin的各种具有挑战性的结构类似物。
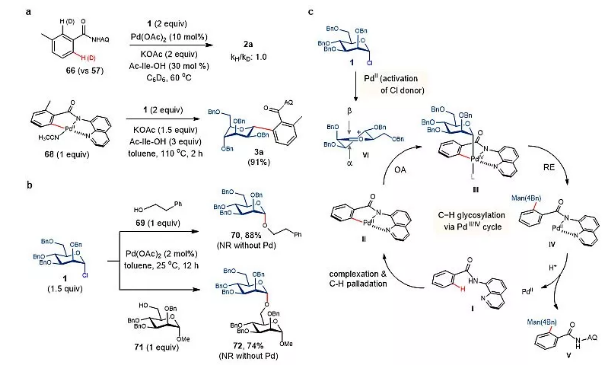
图5. 合成应用。图片来源:Nat. Catal.
最后作者对反应的机理进行了初步探索:如图6a所示,3-甲基苯甲酰胺66和67(kH/kD:1.0)的动力学同位素效应(KIE)实验表明,在AQ导向的Pd-催化循环中,C-H键钯化不是决速步骤。在相同的反应条件B(45 °C)下,66与MeI的邻C-H烷基化反应没有得到邻甲基化产物,说明在官能团化过程中可能存在一种更亲电的物种,而非氯代糖1。大多数氯代糖供体在偶联时立体构型保持,同时使用2,3:4,6-二-O-二异丙基-甘露糖氯供体的2:1 α/β混合物却得到了专一的α-选择性产物23(图2),这表明一种氧鎓正离子中间体可能是活性亲电试剂。如图6b所示,催化量的Pd(OAc)2(2 mol%)在25 ℃时有效地促进了伯醇69和甘露糖受体71与1在甲苯中的糖基化反应形成70和72。如图6c所示,AQ导向糖基化反应的催化循环可能始于底物I的络合和C-H键钯化生成二价钯环中间体Ⅱ。氯供体1被二价钯活化形成甘露糖基氧鎓正离子中间体VI,N,N-双齿AQ配体具有较强的电子供体能力,使Ⅱ的二价钯中心与VI发生氧化加成,形成四价钯中间体Ⅲ,再经还原消除形成IV,IV分解得到最终的糖基化产物V,释放二价钯催化剂。目前还不清楚羧酸添加剂(如HOAc和Ac-Ile-OH)的确切作用。作者怀疑它有助于稳定某些阳离子二价钯物种,它们也有可能作为Lewis酸来活化糖基氯供体,或者作为钯环中间体氧化加成的配体或促进IV分解。然后,C-糖基化与糖基氯的非对映选择性由氧鎓正离子(如VI)的亲电进攻控制,而位阻较大的平面型钯环Ⅱ的空间特征可能是导致亲电进攻得到优异非对映选择性的原因之一。
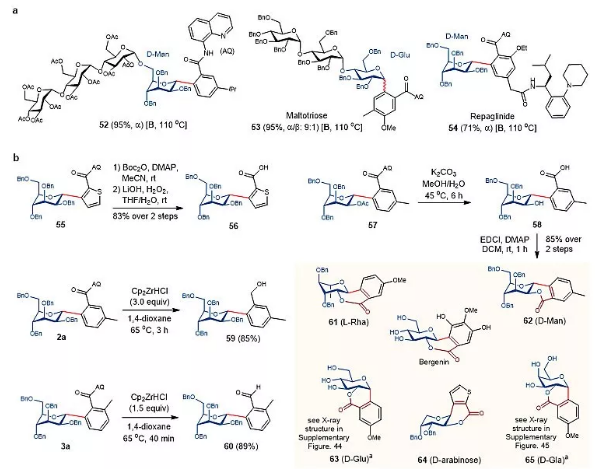
图6. 机理研究。图片来源:Nat. Catal.
总结
南开大学陈弓、何刚课题组发展了一种通过钯催化芳烃和杂芳烃的C-H键糖基化反应合成C-芳基糖苷的策略。该方法效率高,选择性强,适用范围广,操作简单,可以直接合成各种C-芳基吡喃和呋喃糖苷,能够获得现有方法难以制备的高度复杂的产物,为药物分子的后期修饰提供了方法。该反应表现出优异的官能团相容性,并在与糖基氧鎓正离子反应时显示出显著活性。机理研究表明,二价钯作为Lewis酸在活化氯代糖时具有不同寻常的作用。对钯催化的C-H键糖基化反应的深入研究将促进新的C-H键官能团化策略的发现,从而实现复杂 分子的立体选择性合成。
【转载自 X-MOL】

